The immune complement system is a crucial component of the body’s intrinsic immunity, playing roles such as regulating immune responses and clearing pathogens. A growing body of research suggests that the complement system is aberrantly activated in the Alzheimer’s disease (AD) brain and may play a critical role in synaptic loss and cognitive impairment. In various AD mouse models, aberrant activation of complement-dependent synaptic pruning processes causes excessive synaptic clearance, ultimately leading to synaptic loss and impaired learning and memory. This paper summarizes and discusses several key scientific issues in the process of complement-mediated synapse loss in the AD brain.
Sources of Complement Proteins Labeled at Synapses
The current study shows that the levels of complement proteins C1q and C3 are significantly elevated in the brains of AD model mice and AD patients. Animal experiments have shown that complement proteins are mainly derived from microglia and astrocytes. So how do complement proteins of glial cell origin, localize to synapses? The current study suggests at least two possibilities: first, targeted transport via extracellular vesicles such as exosomes; and second, specific binding by recognizing receptors on the synapse. Furthermore, in addition to transfer from glial cells, complement proteins on synapses may also be synthesized locally, i.e., within neurons or synapses. For example, mRNA expression of almost all classical complement pathway proteins (from complement C1q to complement C9) is significantly upregulated in neurons of the brains of AD patients compared with non-AD aged individuals.
Complement Proteins Label Synapses to Trigger Excessive Pruning
Pathological protein components such as Aβ and hyperphosphorylated tau proteins can lead to overactivation of the complement system and consequent deposition at the synapse. Metabotropic glutamate receptors (mGluR1/5), transcription factors, mitochondrial oxidative stress, and apoptotic cascade responses may be involved. For example, it was found that amyloid injection in the CA1 region of the rat hippocampus caused elevated levels of C1q and increased microglia pruning in synaptosomes, ultimately leading to synaptic loss and learning memory impairments; inhibition or knockdown of mGluR1 reversed these phenomena, whereas mGluR1 agonists exacerbated these phenotypes.
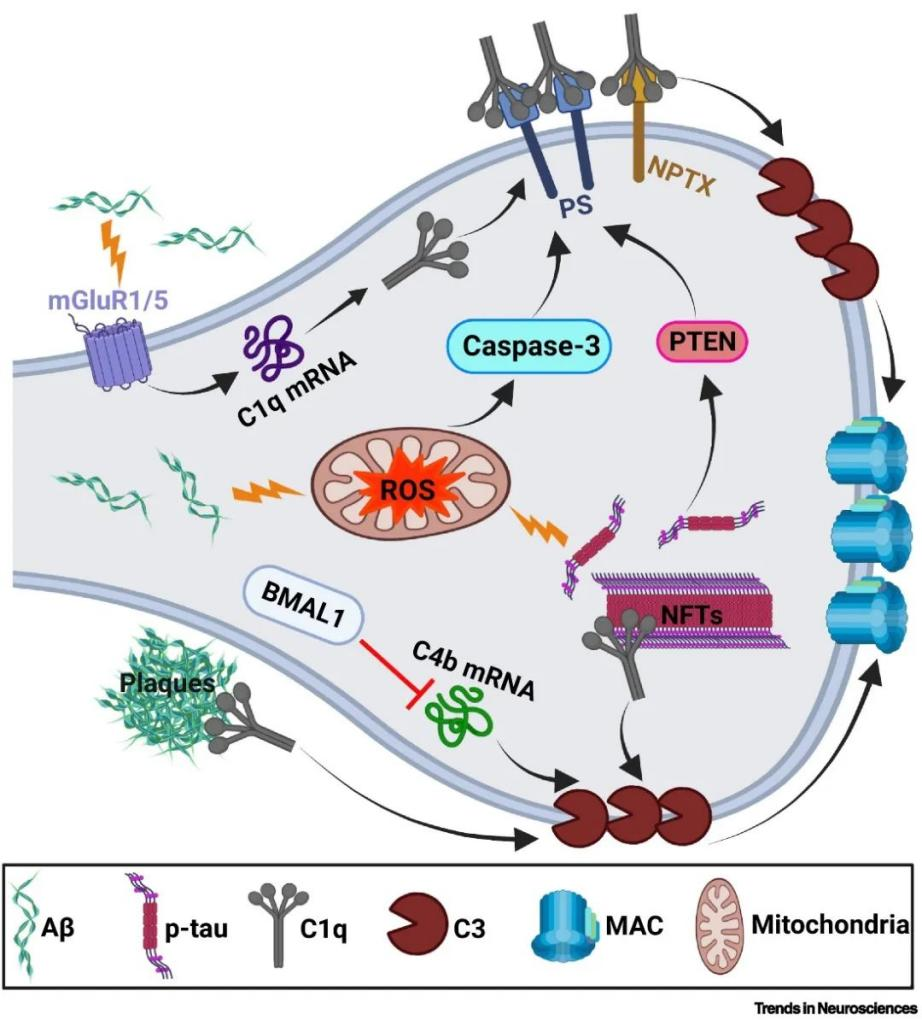
Fig. 1 Factors causing synapses to be labeled by complement.1
How Synapses Labeled by Complement Are Cleared
Complement-labeled synapses in AD are cleared mainly through phagocytosis by glial cells. Phagocytosis of complement-labeled synapses by microglia has been widely reported, while astrocytes can also phagocytose synapses through the MEGF10 and MERTK pathways. In addition, oligodendrocyte precursor cells can also phagocytose synapses, though whether this process is complement-dependent requires further investigation. Molecules secreted by cerebrovascular endothelial cells and pericytes may induce synaptic pruning by microglia. Recent studies have also found that the CD4+ T cell population in the brain is critical for synaptic pruning in microglia. Since blood-brain barrier dysfunction occurs early in AD, peripheral immune cells may infiltrate the brain and modulate complement-dependent synaptic pruning. The effect of these immune cells on complement-dependent synaptic pruning is an important area for future research that could provide new perspectives for understanding the mechanisms of synaptic loss in AD and help develop new therapeutic strategies.
Involvement of AD Risk Factors
Multiple AD risk factors may be involved in complement-mediated synaptic pruning. For example, reports have found that the AD risk gene TREM2, which binds to complement C1q, protects synapses from pruning. Aging, the greatest risk factor for AD, and reduced levels of Progranulin (PGRN) may promote synaptic pruning by increasing the production of C1q in microglia, which in turn exacerbates synaptic loss. Sex differences in AD risk are also evident, with females being more susceptible to AD. This susceptibility is likely related to the dramatic postmenopausal decline in estrogen that leads to increased levels of S-nitrosylated C3 (SNO-C3). The rise in SNO-C3 exacerbates synaptic pruning and synaptic loss in women with AD. In conclusion, there is growing evidence of a possible interaction between the complement system and AD risk factors during synaptic loss. Understanding the role of these risk factors in complement-dependent synaptic pruning will provide new insights into the pathogenesis of AD and potential therapeutic targets.
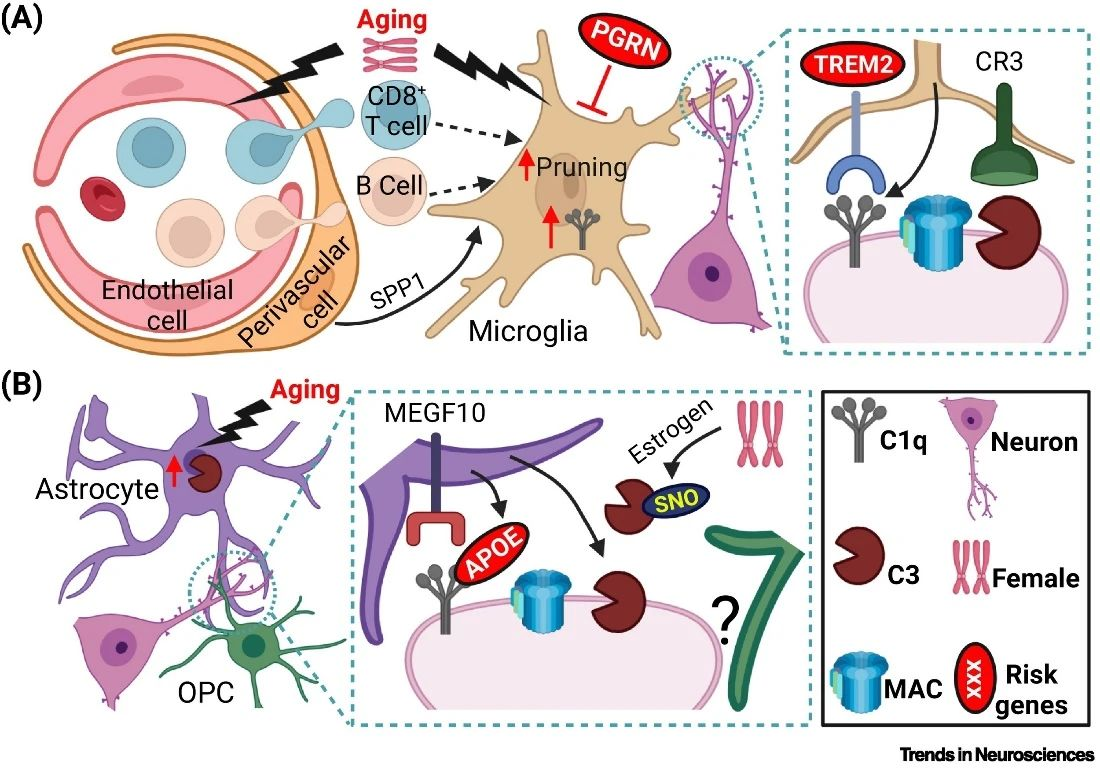
Fig. 2 Different cell types and AD risk factors are involved in complement-mediated synaptic pruning in AD.1
Despite the growing body of evidence from animal experiments supporting complement-mediated synaptic loss in AD, more in-depth studies of specific cellular and molecular mechanisms are needed to develop effective therapeutic treatments. Due to the limitations of animal models in mimicking the complexity of human AD, the use of human cellular models such as induced pluripotent stem cells and brain-like organoids can provide a more comprehensive study of complement-mediated synaptic loss in a context more closely resembling the human brain.
Reference:
- Wen, Lang, Danlei Bi, and Yong Shen. “Complement-mediated synapse loss in Alzheimer’s disease: mechanisms and involvement of risk factors.” Trends in Neurosciences(2024).