Recently, a comment was published in the journal Nature Reviews Materials (impact factor 71.189), outlining the application of extracellular vesicles and synthetic nanoparticles in drug delivery.

Synthetic nanoparticles have been widely used for clinical drug delivery since the 1990s. Designing nano-delivery strategies can improve the spatiotemporal distribution of therapeutic agents in vivo, which can lead to reduced side effects and/or improved therapeutic efficacy. Delivery is improved by optimizing the size, shape, and surface properties of the nanocarriers. However, complex molecular targeting strategies have repeatedly failed in clinical trials. One example is BIND-014, a polymer nanoparticle with surface ligands that binds to prostate-specific membrane antigen (PSMA)-rich tumors. The clinical failure of ligand-targeted nanodelivery approaches has been attributed in part to complex interactions between nanoparticles and the biological environment, including the formation of protein coronas that can mask surface ligands and trigger immunological recognition. This interaction may not be apparent in preclinical studies because major components of the protein corona, such as complement proteins, vary widely between human and animal models. Additionally, ligand type, orientation, density, and surface patterning are critical for optimal delivery and binding to target molecules. Therefore, computational tools are often required to understand how design parameters affect nano-biological interactions.
Most nanomedicines on the market are simple liposomes. Although liposomes are used clinically to deliver a variety of therapeutic agents, including small molecules, peptides, and RNA, the real potential of realizing nano-delivery to improve patient outcomes awaits the realization of more complex multifunctional designs. The optimal design of drug delivery systems may require a level of complexity, similar to that of the biological environment, to overcome various barriers, including clearance, degradation, and physical barriers (e.g., endothelial and cell membranes). Furthermore, site-specific delivery ensures favorable spatiotemporal drug effects. However, it is uncertain whether the successful integration of multiple functional components in synthetic nanodelivery systems can be compatible with cost-effective and time-efficient clinical-grade manufacturing processes.
Extracellular vesicles (EVs) are natural nanoparticles released by cells. EVs are similar in size, shape, and structure to liposomes, but have a more complex bilayer structure containing up to hundreds of different lipids, protein, and carbohydrate types, as well as internal cargo and surface molecules (Figure 1). EVs play important roles in short- and long-distance intercellular communication in various pathological and physiological processes. The ability of EVs to transport biomolecules to recipient cells makes them attractive for drug delivery. EVs can be obtained from conditioned media of cultured cells or from biological tissues or fluids, and various methods such as electroporation, extrusion, and sonication have been used to load therapeutic agents into EVs. By developing EVs for in vivo molecular cargo transport, multiple design and manufacturing challenges of nanomedicine can be circumvented. However, the potential of EVs in drug delivery remains uncertain due to challenges in EV isolation and characterization, which hinders basic and translational research. In particular, the heterogeneity between EV types and the presence of other bionanoparticles with overlapping features make EV isolation difficult. Furthermore, it should be cautious to prematurely describe EVs as a superior drug carrier to synthetic nanoparticles in terms of biocompatibility and site-specific delivery.
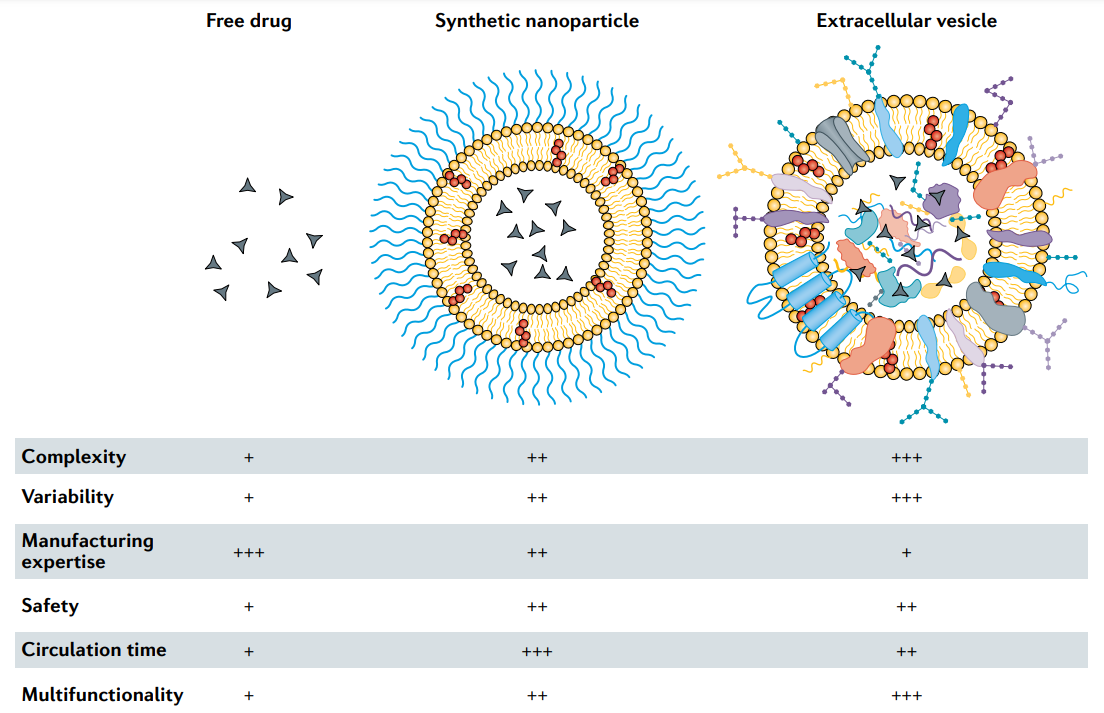
Fig.1 Characteristics of free drugs, clinically approved synthetic nanoparticles and extracellular vesicles. Low (+), medium (++) and high (+++).
Pharmacokinetics
The challenge for nanomedicine is macrophage-mediated hepatic clearance of synthetic nanoparticles from circulation. The clearance of nanocarriers in the liver hinders site-specific delivery, such as tumor targeting of nanocarriers based on size and shape. Many clinically approved synthetic nanoparticles have been functionalized with polyethylene glycol (PEG), which reduces macrophage uptake and extends the circulating half-life from hours to days. The use of PEG also has some disadvantages. EV-based drug delivery could serve as an alternative strategy to avoid immune clearance. However, multiple studies have shown that systemically injected EVs are also rapidly cleared by the liver and have a circulating half-life of only a few minutes. The isolation, drug loading, and labeling procedures of EVs have the potential to make EVs more readily available for rapid uptake by macrophages. However, a study in EV reporter mice showed that cardiac E mainly accumulates in the thymus, testis, lung, and kidney, with less hepatic uptake, suggesting that native EVs formed in vivo may experience less hepatic clearance. Furthermore, it is unclear how EV biogenesis, cellular origin, and biomolecular composition affect pharmacokinetics. Certain EV types have long circulating half-lives and/or have site-specific targeting mechanisms to overcome hepatic clearance. In terms of proper EV isolation, identification, and characterization, experimental design is critical to assess the potential of EVs as drug carriers. Direct comparisons with synthetic nanoparticles are often omitted, however, if some comparisons could be made, this would allow us to determine the benefits of EV delivery in terms of drug toxicity and efficacy.
In addition to biodistribution characteristics that may be superior to synthetic carriers, EV-based drug delivery has another benefit—the possibility to exploit cellular processes for drug loading and surface modification. Cells can be genetically engineered to express and package protein- and RNA-based therapeutics and/or targeting ligands in EVs. Since RNA and proteins may be degraded or destroyed during nanoparticle synthesis, it may be advantageous to utilize cellular machinery for drug loading and EV surface modification. Additionally, EV endocytic pathways or fusion events with recipient cell membranes can facilitate intracellular delivery, i.e., targeting therapeutic agents to specific intracellular compartments or organelles. In a recent study, cells were genetically engineered to enrich for specific small interfering RNAs (siRNAs) in EVs, which resulted in improved delivery of functional siRNAs to mice compared to synthetic lipid nanocarriers more than ten times. The superiority of EV vectors is attributed to the ability of siRNA to escape lysosomes and localize in the cytoplasm. Notably, the efficiency of siRNA-mediated knockdown was dependent on EV source and recipient cell type. Certain cell types, such as resident macrophages, accumulate high levels of EV-delivered siRNA but show minimal target gene knockdown. These results suggest that EV heterogeneity must be considered in terms of biodistribution and intracellular delivery.
Biocompatibility
Nanomedicines generally produce fewer side effects than free drugs because healthy tissue is less exposed to the therapeutic agent and does not require the use of toxic excipients to dissolve water-insoluble small molecules. In fact, several nanodrugs have received clinical approval based on equivalent efficacy but improved safety compared to free-administered small molecules. However, clinically approved nanoparticles containing PEG can activate the complement system and, in rare cases, lead to life-threatening hypersensitivity reactions that are usually alleviated by slowing the infusion rate.
EV-based therapies are not yet clinically approved, but early clinical trials have evaluated the effects of autologous dendritic cell-derived EVs for cancer immunotherapy, and EVs from allogeneic mesenchymal stem cells for regenerative and anti-inflammatory applications. Most of these trials reported mild to moderate side effects, and EV administration is generally considered safe. Allogeneic EVs have been suggested to pose a risk because they may elicit an immune response in the recipient. However, blood and plasma also contain high concentrations of EVs (estimated as high as 1010 EVs per milliliter), which are released into the circulation from all cell types in the body. Indeed, plasma and blood transfusions rarely elicit adverse immune responses, suggesting that allogeneic EVs are unlikely to pose a safety risk. However, the disadvantage of allogeneic EVs compared to autologous EVs may be the accelerated hepatic clearance. Furthermore, although preliminary safety results from clinical trials are encouraging, the challenging endogenous cargo to be removed without damaging EV structures may have adverse consequences. However, endogenous cargo may also contribute to additive or synergistic effects, especially if EVs are obtained from cells with therapeutic properties, such as mesenchymal stem cells.
Outlook
Currently, there are more than 50 clinically approved nanomedicines, all of which are based on simple designs containing few ingredients. The synthesis of more complex nanoparticles with biological structures with hundreds of functional components is unlikely to be compatible with large-scale clinical-scale fabrication. EVs are a promising alternative for realizing multi-pronged and multifunctional drug carriers. In addition to identifying EVs with good delivery properties, challenges in manufacturing and scaling up need to be overcome, including variability in cell culture, which is a major source of EVs. The use of immortalized cell lines minimizes variability, but may present safety concerns because immortalizing agents may be loaded into EVs. There is a further need for large-scale cultures, such as stirred tanks or fixed-bed bioreactors, and chemically defined media or media without xenobiotic supplements, such as human platelet lysates. The effect of lysate material on therapeutic efficacy should be considered. EV mimetics can also be formed by disrupting cells by extrusion or sonication, however, this process may affect membrane topology. Platelets and red blood cells may be particularly suitable sources of EV mimetics because they lack nuclear material, which may act as a red flag for the immune system. Instead of cell culture, human tissues and body fluids can be used as a source of EVs. Indeed, large-scale clinical production of bionanoparticles from human plasma has been demonstrated, for example, for lipoprotein therapy trials with thousands of participants. However, biological fluids and tissues are more complex in composition compared to conditioned cell culture media, making the isolation of EVs from them challenging. For some applications, high-purity EV preparations may not be required, and efficacy may even decrease if cofactors are removed. For example, secretomes obtained from gamma-irradiated peripheral blood mononuclear cells showed higher therapeutic efficacy than EV components alone. In other cases, it may be necessary to separate EVs from components that interfere with EV function and drug loading or even produce deleterious effects. Regulators are still determining which type of standard applies to EV-based products. The presence of other biological components is not necessarily a regulatory barrier, as long as pre-determined physicochemical and potency tests can verify batch-to-batch consistency and efficacy. Finally, non-human EV sources, such as milk, fruits, and algae, have been investigated, but their application is limited due to the potential immune response to intravenous injection.
Therapeutic EVs in clinical trials are typically isolated by ultracentrifugation (both differential and density) and tangential flow filtration. However, most EV-based clinical trials involve only a small number of participants, and ultracentrifugation may not be scalable for larger research and commercial production. Furthermore, ultracentrifugation-based methods may lead to EV damage and aggregation. Tangential flow filtration is compatible with mass production and enables volume reduction and partial decontamination while maintaining EV structure. After purification, EVs can be further engineered by loading with therapeutic agents (clinical-grade protocols have been reported) or by fusing with synthetic nanoparticles. Most clinical trials have used saline or sucrose cryoprotectant buffer to store EVs at -80°C. Alternatively, lyophilization may be suitable for certain EV cargoes and functions and allow for simplified storage, distribution, and use for point-of-care medical services.
The growing dialogue and collaboration between the fields of synthetic nanomedicine and EVs have the potential to drive the successful fusion of emerging EV biology with decades of expertise in the clinical failures and successes of synthetic drug delivery.