Adoptive immunotherapy is a transformational therapy in hematology. One of the most representative adoptive immunotherapies is chimeric antigen receptor (CAR) T cell therapy. It expresses and synthesizes CAR on T cells by genetic engineering. CAR-T cells targeting CD19 antigens in B-cell leukemia and lymphoma have been clinically recognized and have become a “routine” treatment in cancer centers around the world. In other indications of hematology and oncology, great efforts are being made in transformation research to identify and validate new CAR target antigens, improve efficacy and reduce toxicity associated with CAR-T cell therapy, and promote the production of CAR-T cells to increase patient applicability. A series of new targets and techniques are introduced here, which show that researchers are working hard to make CAR-T cell therapy a universal and effective treatment in cancer medicine.
New targets of CAR-T cells
-
Hematological malignant tumor
In hematology, the pipeline with the most abundant new targets is multiple myeloma. Some members of the signaling lymphocytic activation molecule (SLAM) family are being used as potential targets, including SLAMF7 (alias: CD319, CRACC, CS-1) and SLAMF3 (alias: CD229, Ly9). SLAMF7 and SLAMF3 ARE uniformly expressed on tumor cells of untreated and chemotherapy-resistant patients with multiple myeloma. CAR-T cells targeting either antigen showed an efficient killing effect in vitro and in vivo. Therefore, SLAMF7 CAR-T cells are currently entering clinical trials. SLAMF7 is expressed on a small number of normal lymphocyte subsets, resulting in the autophagy of high SLAMF7 expression T cells during the production of CAR-T cells. Therefore, selective loss of SLAMF7 high expression lymphocytes may occur after the adoptive metastasis of SLAMF7 CAR-T cells. It is encouraging that the clinical experience of anti-SLAMF7 antibodies shows that patients retain the total number of lymphocytes without increasing the infection rate. In contrast, the expression of SLAMF3 on T cells, B cells, and NK cells is consistent, indicating that SLAMF3 may lead to lymphocyte depletion, but there is no clinical experience for SLAMF3. The following table provides some other new targeted antigens for CAR-T cells in multiple myeloma, B-cell and T-cell malignant tumors, and acute myeloid leukemia (AML).
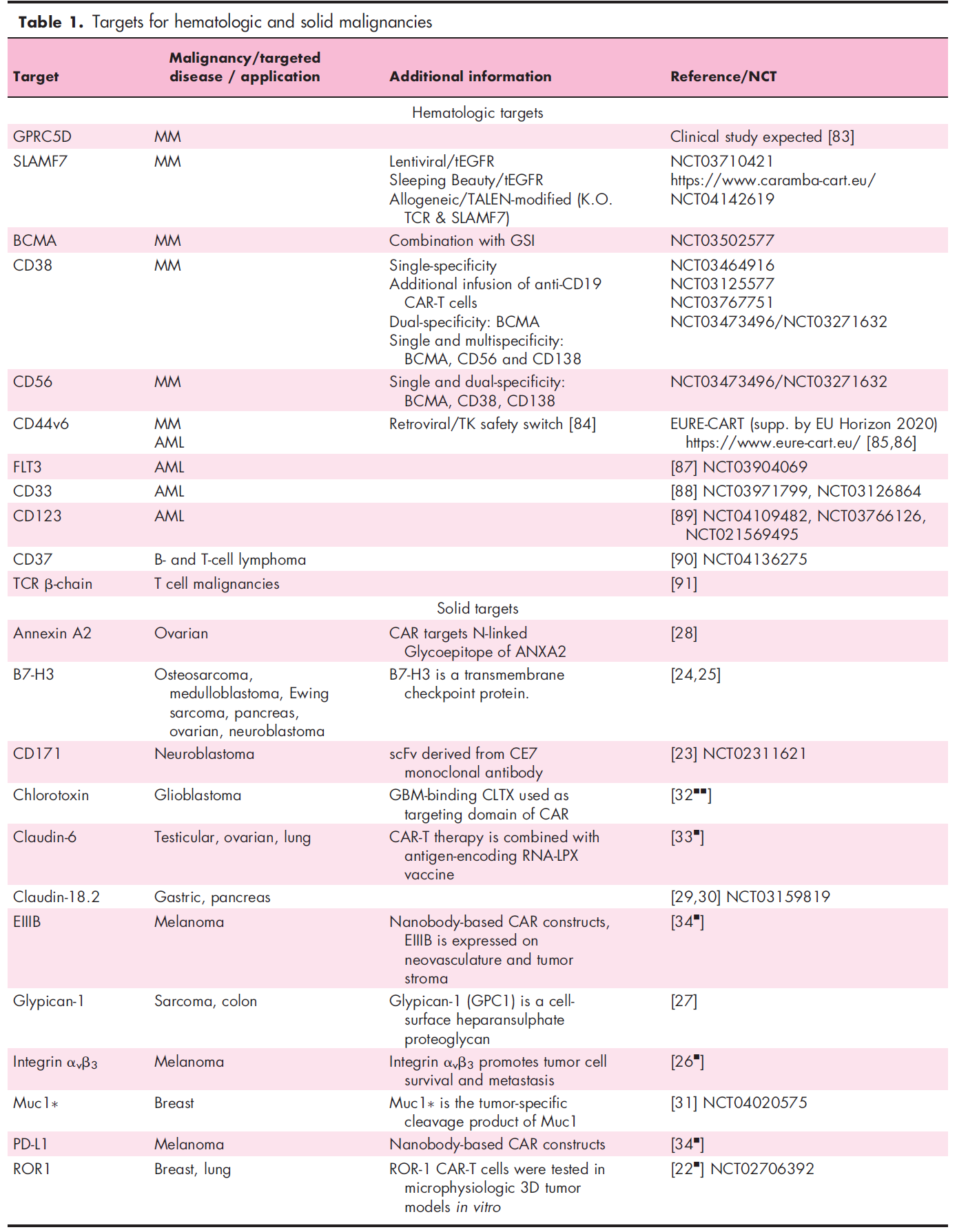
G protein-coupled receptor class C group 5 member D (GPRC5D) has been found to be a gene highly expressed in the bone marrow of patients with multiple myeloma, and is also a factor related to poor clinical prognosis. A recent study showed the expression of GPRC5D protein in primary multiple myeloma cells, and GPRC5D-specific CAR-T cells had an anti-multiple myeloma effect. In addition, the study determined that hair follicles were the only normal tissues that could detect GPRC5D expression. It is worth noting that the combination of targeted B-cell maturation antigen (BCMA) and GPRC5D has been shown to prevent the recurrence of BCMA escape, indicating the potential clinical value of this concept in improving clinical efficacy and prognosis.
Previous studies have reported the clinical efficacy of anti-CD19 CAR-T cells in patients with multiple myeloma, although in clinical pathology, through the routine analysis of flow cytometry, CD19 is rarely detected in multiple myeloma cells. Super-resolution microscopic analysis showed that CD19 was often expressed in primary multiple myeloma cells of some patients, and its molecular density was enough to trigger the recognition of anti-CD19 CAR-T cells. These data suggest that advanced imaging techniques can be used to guide the selection of patients and CAR-T products, as well as to monitor antigen expression during treatment.
Recently, several interesting strategies have been reported to increase the target antigen density of multiple myeloma cells. Small molecule γ-secretase inhibitors (GSIs) have been shown to prevent BCMA shedding from multiple myeloma cells, thus improving the efficacy of anti-BCMA CAR-T cells. It was shown that epigenetic regulation using histone deacetylase (HDAC) inhibitors (e.g., panobinostat, ricolinostat, next-generation HDAC6 inhibitors) can enhance CD38 expression in multiple myeloma cells, thereby enhancing the activity of anti-CD38 antibodies. This concept is currently being tested on targeted antigens of multiple myeloma CAR-T cells.
-
Solid tumor
For a long time, several typical target antigens, such as TAG-72, folate receptor (FR), carcinoembryonic antigen (CEA), GD2, CD171 and interleukin 13 receptor subunit alpha 2 (IL13RA2), have been used to evaluate the therapeutic efficacy of CAR-T cells in solid tumors. Most of these tests use CAR-T products and production processes that are not technologically advanced compared with current standards. Since then, one direction for improving efficacy has been to improve vectors and manufacturing techniques, including the search for alternative target antigens that are expressed homogeneously and stably on tumor cells (ideally with pathophysiological correlation to reduce the risk of antigen loss). At present, several CAR-T cell products are actively undergoing clinical trials or post-clinical development, such as glypican 3 (GPC3), mesothelin, human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor variant III (EGFRvIII), receptor tyrosine kinase like orphan receptor 1 (ROR1), CD171, B7-H3, integrin α v β 3, heparan sulfate Glypican-1, Annexin A2, which connects sugar epitopes at the N-terminal, and MUC1, the cleavage product of Claudin18.2, tumor-restricted MUC1.
In a recent study, researchers discovered the glioblastoma binding ability of scorpion venom chlorotoxin (CLTX) and constructed CAR-T cells with CLTX peptide as the antigen-binding part. The targeting of CLTX-CAR-T cells depends on the expression of MMP2 on the target cells and induces tumor regression without toxicity in the model of xenotransplantation. In addition, there are CAR-T cells targeting Claudin-6 expression in ovarian, testicular and lung cancer. The combined application of RNA vaccine nanoparticles encoding target antigen promoted the expansion and implantation of CAR-T cells in vivo, which led to tumor regression in a homologous mouse model. CAR-T cells are also targeted to non-classical tumor antigens, such as PD-L1 or fibronectin splicing variant EIIIB, which are expressed in tumor stroma and neovascularization.
In addition, another direction is to evaluate the role of CAR-T cells in combination therapy, which aims to neutralize the inhibition mechanism in the tumor microenvironment (TME) and to use advanced genetic engineering techniques to enhance the efficacy and lifespan of CAR-T cells.
New techniques of CAR-T therapy
-
Techniques to improve efficiency
T cell failure has been identified as a major reason for limiting the efficacy of CAR-T cells in the treatment of solid tumors. In order to construct anti-failure CAR-T cells, researchers recently identified key transcription factors and showed that the deletion of TOX and NR4A transcription factors and the overexpression of AP-1 family transcription factors c-Jun increased the function of CAR-T cells in mouse tumor model.
In TME, many inhibitors may hinder the function of CAR-T cells. Anti-PD1 scFv produced by CAR-T cells, removal of PD-1 and TGF- β receptors, inhibition of TGF- β receptor signaling pathway and expression of the main negative regulation of TGF-β receptors can resist these inhibitory ligands and soluble factors, thus enhancing the function of CAR-T cells. In addition, the death signal from TME can be reduced by expressing the main negative Fas receptor in CAR-T cells.
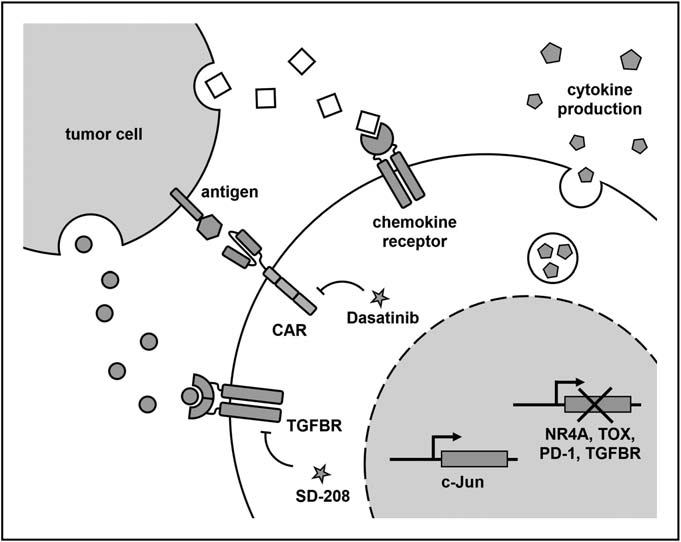
Guiding CAR-T cells to actively regulate the cytokine environment in TME is another effective strategy to enhance efficacy. Recently, it has been reported that making CAR-T cells produce IL-15, or the combination of IL-15 with IL-21 or IL-23 can improve the persistence and anti-tumor ability of CAR-T cells through an autocrine mechanism. Similar effects were observed on IL-18-producing CAR-T cells or oncolytic virus targeting the delivery of IL-2 and TNF-a to tumors, both of which had the additional effect of participating in congenital and endogenous adaptive immune responses. Similarly, the expression of CD40L on CAR-T cells can make antigen presenting cells recruit endogenous tumor targeting T cells.
The decrease of metastasis of CAR-T cells to the tumor site may be caused by the mismatch of chemokine receptors on CAR-T cells and the abnormal expression of chemokines in tumors. Therefore, in order to promote tumor delivery, CAR-T cells can be transformed into chemokine receptors that match the chemokine profile of a specific tumor. The expression of CCR2b allows CAR-T cells to migrate to tumors expressing CCL2. Similarly, a recent study combined the expression of IL-8 receptors with CXCR1 or CXCR2 to increase the persistence and delivery of CAR-T cells to tumor sites. In addition, it is conceivable that some of these strategies can be combined to “customize” CAR-T cell products to suit the specific circumstances of each tumor entity.
-
Techniques to improve safety
Cytokine release syndrome (CRS) is a systemic inflammatory response and the most common acute toxicity of CAR-T cell therapy. Several factors affect the incidence and severity of CRS, including tumor load, CAR-T cell dose, in vivo expansion and regulation of lymphocyte consumption. The clinical management concepts of CRS and neurotoxicity continue to be refined and contribute to the reduction of morbidity and mortality. However, CRS remains the main cause of hospitalization and intensive care, limiting the availability of CAR-T cell therapy.
An important recent development is the development of mouse models that can reproduce the clinical characteristics and neurotoxicity of CRS and are used to gain an in-depth understanding of the pathophysiology of CRS and to evaluate intervention strategies. In the NSG-SGM model, mice are implanted with human hematopoietic stem cells to provide “endogenous” myeloid cells that play an important role in the vicious cycle of CRS development and produce CRS dominant cytokines IL-6 and IL-1. In this model, the use of the clinically approved IL-6 receptor antagonist can prevent the development of dominant CRS, but does not prevent neurotoxicity. This provides the hypothesis that IL-1 secretion is a key event in the occurrence of CRS. In fact, the treatment of IL-1 receptor antagonist anakinra can control CRS, and prevent fatal neurotoxicity. Phase II studies of the use of Anakinra in CD19 CAR-T therapy to prevent CRS and neurotoxicity are ongoing. Similarly, in the mouse model of xenotransplantation, the combination of anti-GM-CSF antibody lenzilumab enhanced the anti-tumor activity of CAR-T cells and reduced CRS and neuroinflammation.
In addition, it was found that dasatinib, an inhibitor of tyrosine kinase (TKI), induced rapid startup and shutdown of CAR-T cells by blocking LCK interference with CAR signal, which was characterized by cytolytic activity and cessation of cytokine secretion. To demonstrate the ability to “remotely control” CAR-T cells in vivo, the researchers used dasatinib as a pharmacological switch to prevent the secretion of cytokines and the development of CRS in mice. Interestingly, the inhibitory effect of dasatinib is rapid and completely reversible, providing an opportunity for acute and transient control without terminating CAR-T products, as in the case of triggering a suicide switch (e.g. inducible caspase 9), or antibody-based clearance by co-expressing surface markers such as tEGFR.
Another recently proposed strategy is to design a brake structure that contains heterodimer target recognition and signal domains that can be destroyed when using high-affinity small molecular drugs. In the presence of drugs, the brake structure is inactivated. One advantage is that the small molecule drug specifically targets this particular brake structure, while the disadvantage is that the drug still needs to be formally developed and approved clinically. In addition to temporary toxicity control, these two strategies can also be coordinated to prevent chronic CAR signaling, which is a key factor leading to CAR-T cell failure.
There is an increasing number of studies on improving the ability of CAR-T cells to distinguish tumor cells from healthy cells by expressing “intelligent” CAR designs that take into account multiple antigens. An advanced concept is the use of synthetic Notch (SynNotch) receptors, which contain extracellular antigen recognition domains that target specific targets. The target junction leads to the release of intracellular transcription factor, which migrates to the nucleus to induce the expression of the secondary CAR gene. In a recent application, B7-H3 or EpCAM-specific SynNotch receptors were used to induce the expression of ROR1-specific CAR. This study shows the potential to improve tumor cell selectivity and prevent toxicity to healthy tissues. However, the concept of SynNotch is still plagued by the spontaneous release of notch receptors and the time delay in the expression of secondary CAR on T cells. The effectiveness and safety of SynNotch CAR-T cells have yet to be clinically proved.
-
Improvement of Genetic Engineering and Manufacturing Technology
Two recent developments that are particularly noteworthy are the use of the Sleeping Beauty transposon system and CRISPR-Cas9 genome editing technology to improve virus-free gene transfer and targeted genome insertion of CAR transgenes. The development of design variants of Sleeping Beauty transposons can co-transform T cells through a small cycle of DNA transposons, thus promoting scalable, rapid and exportable manufacturing in a virus-free process. Previous studies have demonstrated the potential for stable CAR expression and established the first clinical safety study for Sleeping Beauty CAR-T cells. Europe’s first clinical trial of Sleeping Beauty CAR-T cells has recently been approved and will study the efficacy of anti-SLAMF7 CAR-T cells against multiple myeloma. The CAR (and T cell receptor, TCR) gene is targeted to insert into the endogenous TCR site in order to achieve physiological regulation of CAR expression to enhance responsiveness and prevent depletion. A key requirement for clinical use of CRISPR-edited T cell products is to carefully annotate non-targeted genes to assess the risk of genotoxicity.
Prospect
In hematology and oncology indications, the new target antigens of CAR-T cells are rich in pipelines, and there are also new techniques to improve the therapeutic index of CAR-T cell therapy. With this “goal and technology package”, we have reason to expect CAR-T cell therapy to complete its conceptual clinical practice in other cancer entities.
Reference
1. Freitag, Fabian, et al. “New targets and technologies for CAR-T cells.” Current Opinion in Oncology 32.5 (2020): 510-517.