DCR-PH1
DCR-PH1 for Primary Hyperoxaluria Type 1 (PH1)
DCR-PH1, an investigational substrate short interfering RNA (siRNA) therapeutic, is a proprietary therapeutic candidate in development for the treatment of PH1 by targeting the liver metabolic enzyme glycolate oxidase (GO). DCR-PH1 is designated as Orphan Drug in the U.S. Food and Drug Administration (FDA) and European Union (EU) in April and August 2015. PH1 is a severe, rare, inherited disorder of the liver which causes overproduction of oxalate due to a genetic mutation in the AGXT gene encoding the glyoxylate-aminotransferase (AGT). These often lead to the deposition of calcium oxalate crystals in the kidneys and further result in severe damage to the kidneys and other organs, eventually kidney failure. Currently, there are no approved therapies for the treatment of PH1, only the liver-kidney transplants as the major surgical procedures which are often difficult to perform with substantial associated risks. This DCR-PH1 as one of the siRNA-based therapeutics offers a new treatment option for rare and hard-to-cure diseases like PH1.
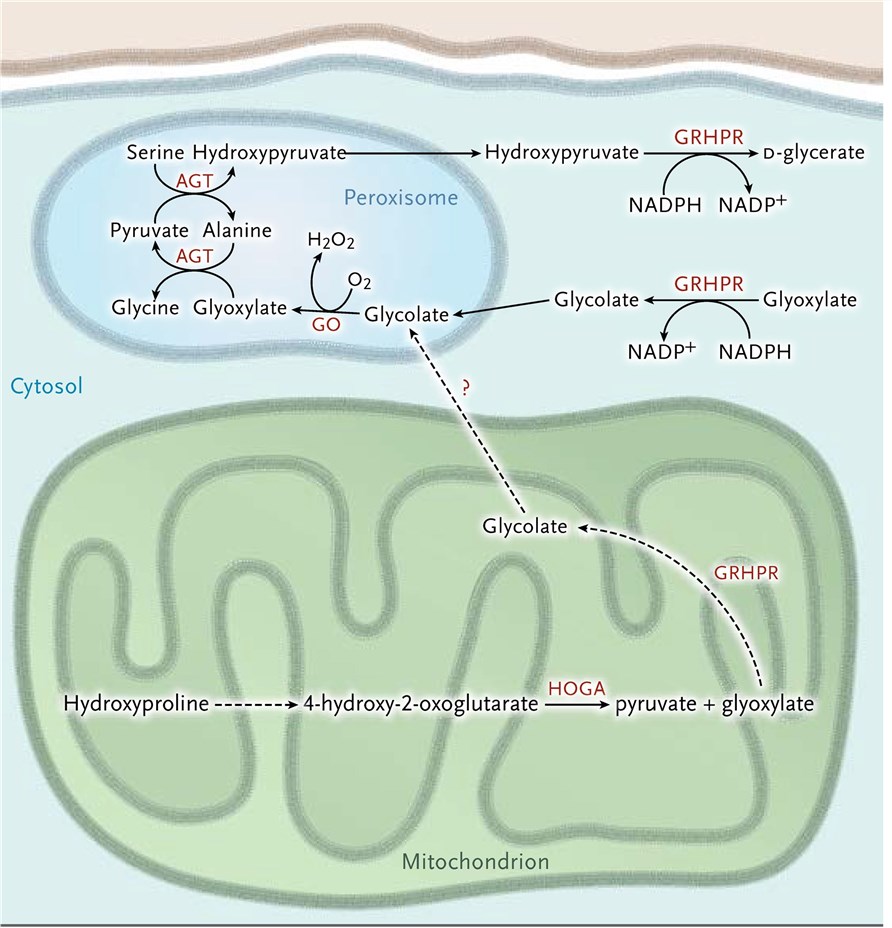 Figure 1. Glyoxylate metabolism in the hepatocyte. (Cochat, 2013)
Figure 1. Glyoxylate metabolism in the hepatocyte. (Cochat, 2013)
Mechanism and Preclinical Study of DCR-PH1
DCR-PH1 is designed to target and destruct the messenger RNA (mRNA) produced by the HAO1 gene encoding GO, an upstream enzyme involved in the production of oxalate. GO is implicated in the pathogenesis and progression of PH1. DCR-PH1 could reduce oxalate production in order to address the pathology of PH1 and prevent the complications of PH1. DCR-PH1 is processed by the Dicer enzyme, which is the initiation point for RNA interference (RNAi) in the human cell cytoplasm to maximize RNAi potency. Moreover, DCR-PH1 incorporates siRNA formulated in a proprietary lipid nanoparticle (LNP) technology that is an effective system for delivery to the liver after intravenous (IV) administration.
Preclinical studies in a genetic mouse model of PH1 have indicated that DCR-PH1 induces potent and long-term inhibition of HAO1 encoding GO and reduces the production of oxalate levels to near normalization to significantly eliminate the key PH1 disease pathology. In animal models of PH1, DCR-PH1 also significantly increases the levels of glycolate, a metabolite that is the substrate of the GO enzyme, while demonstrating the long-term efficacy and tolerability.
| Clinical Trial | Study | Conditions | Status |
| Phase 1 | A study of DCR-PH1 in patients with primary hyperoxaluria type 1 (PH1) | PH1 | Terminated |
DCR-PH1 into Clinical Trials
Given the encouraging inhibitory activity of DCR-PH1 in animal studies, DCR-PH1 has studied in two Phase 1 clinical trials to dosing in humans: DCR-PH1-101 in patients with PH1 and DCR-PH1-102 in normal healthy volunteers. The DCR-PH1-101 clinical trial tests single ascending doses of DCR-PH1 in patients. Investigators monitor patients for changes in urinary and plasma glycolate and oxalate, key efficacy markers in PH1. It is shown that DCR-PH1 is well tolerated up to a dose of 0.05 mg/kg. The clinical trials are demonstrated that the glycolate levels in the urine are increased in the dosing groups which is consistent with the pharmacological activity of DCR-PH1 in blocking the oxalate pathway. These results provide RNAi proof-of-concept for the mechanism of action of DCR-PH1 targeting GO; however, DCR-PH1 clinical trial programs are now discontinued, leading to the development of the novel GalXC product candidates.
Although the development program of DCR-PH1 has discontinued, it also provides the encouraging results and the important justification to expect for RNAi therapy as the innovative approach to treat PH1. Creative Biolabs is proud to provide a wealth of opinion and experience of the siRNA-based therapeutics, please feel free to contact us if you are interested.
Reference
- Cochat, P.; Rumsby G. (2013). Primary hyperoxaluria. New England Journal of Medicine. 369(7): 649-658.