Overview of Autosomal Recessive Disorders
Autosomal recessive disorders are a class of genetic disorders that require the inheritance of a mutation in the same gene from both parents to occur. These disorders are characterized by genes located on the non-sexed part of the chromosome and are recessive, meaning that only two genes are abnormal to cause the disorder. Common autosomal recessive disorders include cystic fibrosis, sickle cell anemia, Tay-Sachs disease, phenylketonuria, thalassemia, albinism, galactosemia, Spinal muscular atrophy, and others. A variety of autosomal recessive disorders will have a serious impact on the lives of children, and, in some cases, may even be fatal. Therefore, screening is necessary for those who are at high risk of carrying the mutated gene or who want to know about their fertility risks.
Cystic Fibrosis
Cystic fibrosis is an autosomal recessive genetic disease caused by mutations in the CFTR gene, which mainly affects the lungs and digestive system. This mutation causes the secretions (such as mucus, sweat, and digestive fluids) to become thick and sticky, blocking the tubes, ducts, and passages and causing various complications. The symptoms of cystic fibrosis include persistent coughing, coughing up thick mucus, shortness of breath, repeated lung infections, nasal inflammation or congestion, recurrent sinusitis, pancreatitis, difficulty gaining weight, high salt content in sweat and skin, heat intolerance, fertility problems, and arthritis. Cystic fibrosis is usually diagnosed by a newborn screening test (heel prick test), which can detect more than 95% of babies with CF. If the test result is positive, then the baby should have a sweat test at 6 weeks old to determine if they have CF or are healthy carriers of the CF gene. Adults who have a family history of CF can consult a genetic expert and have blood tests to determine if they carry the CF gene.
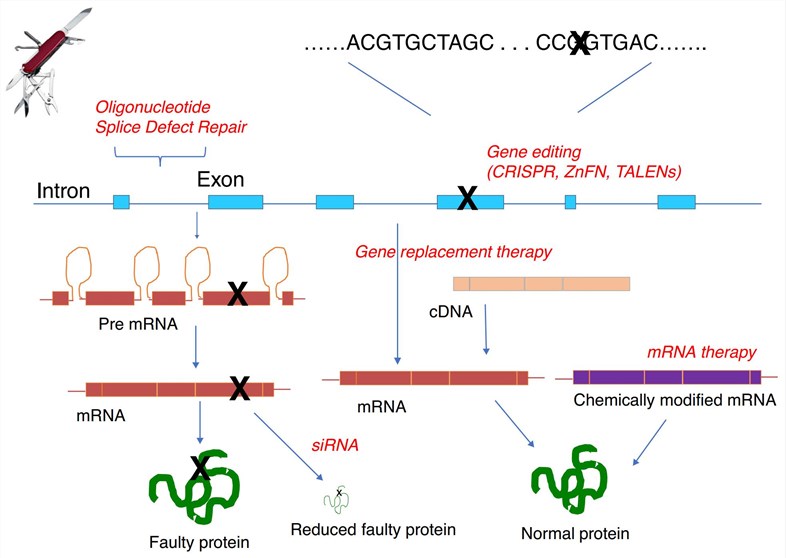 Fig.1 Therapeutic strategies for gene therapy of cystic fibrosis (Hart 2017)
Fig.1 Therapeutic strategies for gene therapy of cystic fibrosis (Hart 2017)
There is no cure for cystic fibrosis, but there are some treatments to relieve symptoms and prolong life. Cystic fibrosis treatment usually includes physiotherapy every day to clear the lungs of fluid, taking capsules to supplement digestive enzymes to help digest food, taking antibiotics to prevent or treat lung infections, using inhalers to dilate the airways, supplementing salt and vitamins, and following a special diet. Some CF patients may need a lung transplant to prolong their lives. Others may need a liver or pancreatic transplant. Gene therapy is a potential way to cure cystic fibrosis and aims to repair or replace the defective CFTR gene, thereby restoring normal secretion function. Currently, there is no clinically validated gene therapy available for CF patients, but there are some experimental trials and studies underway.
Sickle Cell Anemia
Sickle cell anemia is an inherited blood disorder that primarily affects people of black or African descent. Affected patients have sickle-shaped red blood cells that cannot carry oxygen properly and are easily destroyed, leading to anemia, pain, infection, and organ damage. Sickle cell anemia is caused when both parents carry an abnormal hemoglobin gene. The diagnosis of sickle cell anemia relies on blood tests that can detect abnormal hemoglobin and sickle-shaped red blood cells. The treatment of sickle cell anemia mainly includes drug therapy, blood transfusion therapy, and bone marrow transplantation. Medical treatment mainly uses drugs such as hydroxyurea to increase normal hemoglobin and reduce sickle red blood cell formation. Transfusion therapy is primarily used to improve oxygen delivery and reduce the severity of anemia. Bone marrow transplantation is the only method that can cure sickle cell anemia, but there are problems such as donor matching, rejection, and long-term complications. At present, gene therapy is an emerging method for the treatment of sickle cell anemia, mainly by modifying the patient's own hematopoietic stem cells so that they can produce normal or functional hemoglobin. This approach promises to avoid the risks and limitations of bone marrow transplantation, but more clinical trials and safety assessments are still needed.
Tay-Sachs Disease
Tay-Sachs disease is a genetic disorder in which a deficiency of an enzyme called hexosaminidase A causes the abnormal accumulation of a lipid called GM2 ganglioside in nerve cells, which destroys the nervous system. There are two main types of this disease: infantile and late-onset. The infantile form is the most common, with symptoms usually starting 3-6 months after birth and including loss of the ability to turn over, sit, or crawl and red spots on the retina. Seizures, hearing loss, movement disorders, and other symptoms follow, and death usually occurs before the age of 5. Late onset is less common and can appear in adolescence or adulthood with milder symptoms, but it also causes cognitive and motor decline. There is currently no cure for Tay-Sachs disease, only supportive care and psychological support are available. However, there are gene therapy options under investigation that aim to reduce the accumulation of GM2 ganglioside by delivering the normal HEXA gene to nerve cells to restore the enzyme's activity. For example, one gene therapy protocol uses a modified adeno-associated virus (AAV) as a vector to inject the HEXA gene into the cerebrospinal fluid. This regimen has shown some effect in mouse and monkey models, but further clinical trials are needed to prove its safety and efficacy.
Phenylketonuria
Phenylketonuria (PKU) is a rare inherited metabolic disorder that causes the amino acid phenylalanine to accumulate in the body. PKU is caused by mutations in the phenylalanine hydroxylase (PAH) gene. This gene helps produce the enzyme needed to break down phenylalanine. Without the enzymes needed to break down phenylalanine, a dangerous buildup of phenylalanine can result when people with PKU ingest foods that contain protein or contain the artificial sweetener aspartame. This can eventually lead to serious health problems. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It can also cause breath, a musty smell in the skin or urine, and lighter skin, hair, and eye color in family members because phenylalanine cannot be converted into melanin, the pigment that determines hair and skin tone. If a woman with untreated PKU becomes pregnant, it can lead to fetal heart problems, a small head, and low birth weight. The diagnosis of PKU relies primarily on newborn screening programs that are implemented in many countries.
PKU is treated with a low-protein diet that restricts phenylalanine and supplements with specific nutrients. This diet should be started as soon as possible after birth and maintained throughout life. Early diagnosis and maintenance of a strict diet can prevent intellectual disability and other complications, and maintain normal health and lifespan. Treatment effects are monitored with regular blood tests. Medications such as saprotriptyline dihydrochloride or pervalis may help some people. Currently, there are no gene therapy options for PKU, but there are ongoing or planned clinical trials aimed at repairing or replacing the PAH gene or enzyme through methods such as gene editing or gene transfer. These regimens are still in their early stages, and more research and evaluation are needed to determine their safety and effectiveness.
Thalassemia
Thalassemia is an inherited blood disorder that causes excessive destruction of red blood cells and anemia due to abnormal synthesis of hemoglobin. Thalassemia occurs due to mutations or deletions in genes required for hemoglobin synthesis. Hemoglobin is composed of two globins, α and β. If the α-globin gene is missing or abnormal, α-Thalassemia will result. β-Thalassemia occurs when the gene for β-globin is missing or abnormal. Both types of Thalassemia have different subtypes and differ in severity. Symptoms of thalassemia also vary by type and severity, and some people may have no obvious symptoms, while others have facial bone deformities, dark urine, growth retardation, fatigue, pale or yellow skin, and enlarged spleen and other symptoms. Thalassemia is diagnosed primarily through blood tests, including a complete blood count, special hemoglobin tests, and genetic testing. The type and subtype of Thalassemia can be determined by observing whether the morphology of red blood cells is abnormal, and performing hemoglobin electrophoresis. In some cases, Thalassemia can also be diagnosed through prenatal testing.
Treatment options for Thalassemia depend on the type and severity. For more severe cases, treatments such as regular blood transfusions, iron chelators, folic acid, etc. are usually required. Blood transfusions can raise hemoglobin levels, but they can also lead to iron accumulation in the body, so iron chelators are needed to flush out excess iron. Folic acid can help make new red blood cells. There are different types of iron chelators, such as deferoxamine, defesrol, or deferiprone. In some cases, a bone marrow transplant may also be considered. Gene therapy is an emerging treatment for Thalassemia that restores normal hemoglobin synthesis by repairing or replacing the abnormal gene. There have been some successful cases, such as the use of CRISPR-Cas9 technology to edit the stem cell genes of patients with β-Thalassemia so that they can produce fetal hemoglobin (HbF), thereby reducing the dependence on blood transfusions. There are also ongoing clinical trials, such as the use of retroviral vectors to transfect normal beta globin genes into a patient's own stem cells, which are then transplanted back into the patient. Gene therapy also faces some challenges and risks, such as safety, effectiveness, cost, ethics and other issues.
References
- Rafeeq MM, et al. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017 Apr 27;15(1):84.
- Polgreen PM, et al. Clinical Phenotypes of Cystic Fibrosis Carriers. Annu Rev Med. 2022 Jan 27;73:563-574.
- Williams TN, et al. Sickle Cell Anemia and Its Phenotypes. Annu Rev Genomics Hum Genet. 2018 Aug 31;19:113-147.
- Steinberg MH. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. ScientificWorldJournal. 2008 Dec 25;8:1295-324.
- Kavanagh PL, et al. Sickle Cell Disease: A Review. JAMA. 2022 Jul 5;328(1):57-68.
- Solovyeva VV, et al. New Approaches to Tay-Sachs Disease Therapy. Front Physiol. 2018 Nov 20;9:1663.
- Picache JA, et al. Therapeutic Strategies For Tay-Sachs Disease. Front Pharmacol. 2022 Jul 5;13:906647.
- Hussein N, et al. Preconception risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay-Sachs disease. Cochrane Database Syst Rev. 2021 Oct 11;10(10):CD010849.
- Wiedemann A, et al. Phenylketonuria, from diet to gene therapy. Med Sci (Paris). 2020 Aug-Sep;36(8-9):725-734. French.
- Blau N. Genetics of Phenylketonuria: Then and Now. Hum Mutat. 2016 Jun;37(6):508-15.
- Lichter-Konecki U, et al. Phenylketonuria: Current Treatments and Future Developments. Drugs. 2019 Apr;79(5):495-500.
- Viprakasit V, et al. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol Oncol Clin North Am. 2018 Apr;32(2):193-211.
- Motta I, et al. Beta Thalassemia: New Therapeutic Options Beyond Transfusion and Iron Chelation. Drugs. 2020 Jul;80(11):1053-1063.
- Hart, Stephen L., and Patrick T. Harrison. "Genetic therapies for cystic fibrosis lung disease." Current Opinion in Pharmacology 34 (2017): 119-124.