Overview of X-Linked Dominant Disorders
X-linked dominant disorders are a group of genetic diseases caused by dominant mutations on the X chromosome. This means that having one copy of the mutated X chromosome is enough to cause the disease. The X chromosome has many important genes that control various aspects of human development, metabolism, immunity, and the nervous system. If these genes are mutated, they will affect normal physiological functions and phenotypic traits. The severity and manifestation of X-linked dominant disorders vary from person to person depending on the type, location, size, and functional impact of the mutation. Some mutations may cause embryonic lethality or early death; some mutations may cause mild or hidden abnormalities; and some mutations may cause damage to different organs or systems. In addition, since females have two X chromosomes, one of them will be randomly inactivated in each cell, forming a Barr body. This random inactivation will cause different cells to express different X chromosomes, resulting in different degrees of impact. This phenomenon is called heterogeneity or mosaicism. Common X-linked dominant disorders include Incontinentia pigmenti, hypophosphatemic rickets, Aicardi syndrome, Goltz syndrome, Cornelia de Lange syndrome, Alport syndrome, and others. These diseases are all caused by dominant mutations on the X chromosome, involving abnormalities in the nervous system, skin, bones, eyes, ears, a kidney, and development.
Incontinentia Pigmenti
Incontinentia pigmenti is a rare X-linked dominant genetic disorder that affects the skin, eyes, teeth, nails, and central nervous system. It gets its name from its appearance under a microscope. The condition is characterized by skin abnormalities that begin in childhood, usually a blistering rash that heals to form firmer skin growths. Gray or brown patches of skin may appear that fade over time. Other symptoms may include hair loss, dental abnormalities, eye abnormalities (which may lead to vision loss), and textured or pitted nails. Associated problems may include developmental delays, intellectual disabilities, epilepsy, and other neurological problems. Most males who carry the gene for the disease do not survive to be born. TNF-alpha-induced apoptosis. Lack of IKBKG thus predisposes cells to apoptosis. In the skin, cell death may manifest as blisters. These blisters heal after the cells carrying the mutated gene die and are replaced by surrounding cells. Cell death also affected endothelial cells in the brain. This leads to the formation of abnormal blood vessels and the leakage of proteins from the blood into the brain. This can cause seizures. The cutaneous manifestations of Incontinentia pigmenti have four known clinical stages, but they are in an irregular sequence, of variable duration, and may overlap. Treatment options for Incontinentia pigmenti are aimed at the management and prevention of various complications, including skin, eye, dental, nervous system and psychological problems. No specific gene therapy options are currently available.
Hypophosphatemic Rickets
Hypophosphatemic rickets is a genetic disorder characterized by low levels of phosphate in the blood, leading to bone softening and rickets or osteomalacia. It is usually caused by gene mutations that affect the kidney’s reabsorption of phosphate and the intestine’s absorption of calcium. It has several forms, the most common being X-linked hypophosphatemia, caused by mutations in the PHEX gene. The symptoms of hypophosphatemic rickets usually appear in childhood and vary in severity. Severe forms may cause leg bowing and other bone deformities, bone pain, joint pain, poor bone growth, and short stature. Some patients may also have premature closure of the skull sutures (craniosynostosis), leading to developmental abnormalities. The diagnosis of hypophosphatemic rickets mainly relies on the levels of serum phosphate, alkaline phosphatase, and 1,25-dihydroxyvitamin D3, as well as the excretion of calcium and phosphate in urine. Sometimes additional tests such as X-rays, genetic testing, and kidney biopsy may be needed.
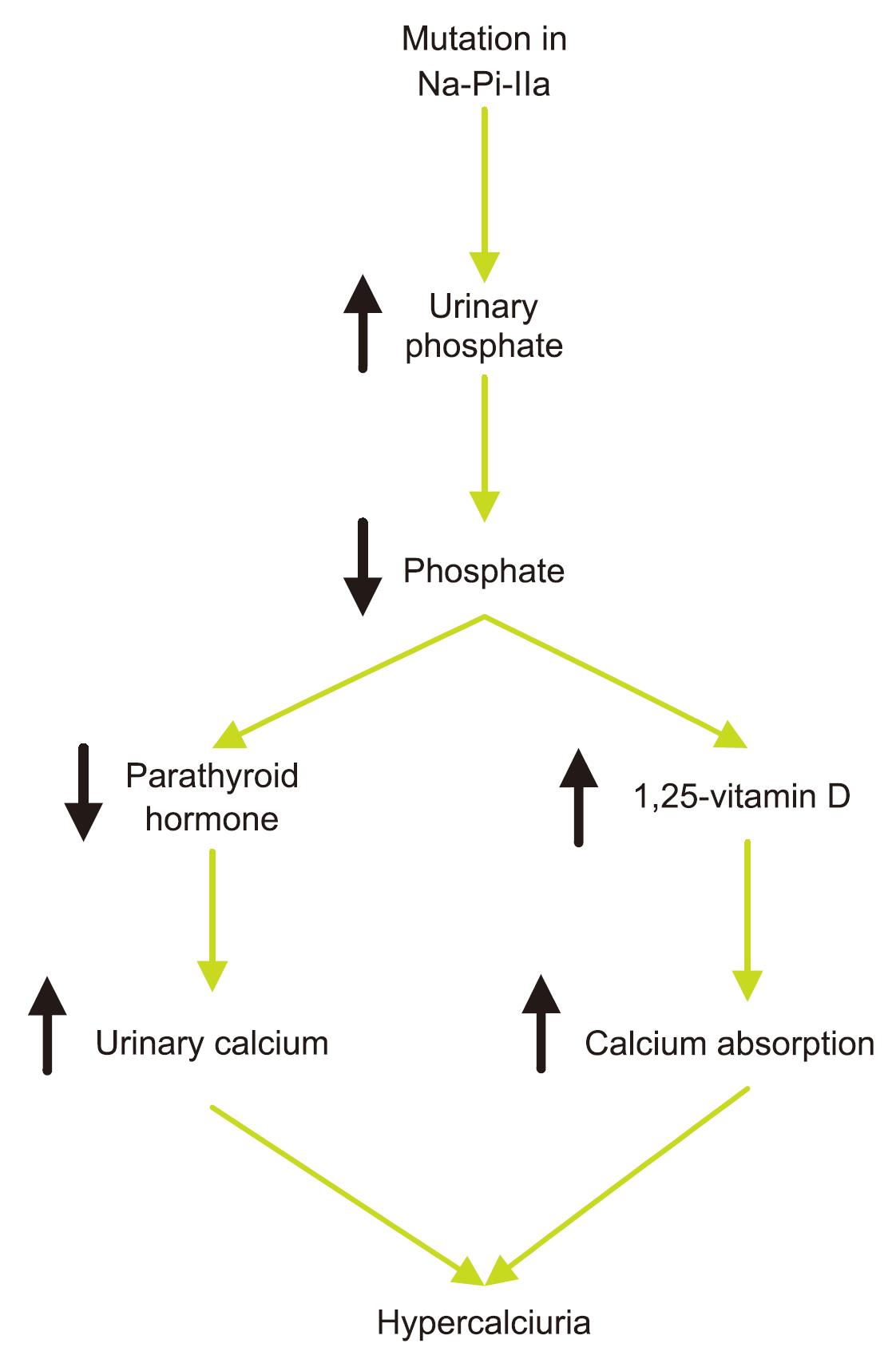 Fig.1 Pathogenesis of Hereditary Hypophosphatemic Rickets with Hypercalciuria. (RASTGAR,2009)
Fig.1 Pathogenesis of Hereditary Hypophosphatemic Rickets with Hypercalciuria. (RASTGAR,2009)
The treatment of hypophosphatemic rickets mainly consists of oral phosphate and calcium supplements, as well as vitamin D analogs (such as calcitriol). These drugs can increase serum phosphate levels, promote bone mineralization, and improve bone deformities and growth development. However, these drugs also have some side effects, such as hypercalciuria, kidney stones, and soft tissue calcification. Currently, there is no effective gene therapy that can cure hypophosphatemic rickets. But there are some novel drugs under development, such as burosumab (KRN23), which is a monoclonal antibody that inhibits the activity of FGF-23. FGF-23 is a hormone that regulates phosphate metabolism and is overproduced in hypophosphatemic rickets, causing the kidney to excrete too much phosphate. Burosumab can lower FGF-23 levels, increase serum phosphate levels, and improve bone health. Currently, burosumab has been tested in clinical trials in children and adults with X-linked hypophosphatemia and has shown some efficacy.
Aicardi Syndrome
Aicardi syndrome is a rare genetic neurological disorder characterized by the partial or complete absence of a key structure in the brain called the corpus callosum, which connects the two halves of the brain, the presence of retinal lacunes, and epileptic seizures in the form of infantile spasms. This disorder primarily affects newborn girls and the precise gene or gene mechanism that causes it has not been identified. Neurological symptoms of Aicardi syndrome may include infantile spasms (a type of seizure disorder), retinal lacunes, lower muscle tone around the head and trunk of the body, microcephaly (small head circumference), and others. People with Aicardi syndrome may also have developmental abnormalities of the optic nerve, small eyes, skeletal problems (such as absent or abnormal ribs and abnormalities of vertebrae in the spinal column), skin problems, facial asymmetry, small hands, and an increased incidence of tumors. There is no cure for Aicardi syndrome and treatment is symptomatic, such as managing seizures and helping parents and children cope with developmental delays. Depending on its severity, Aicardi syndrome can be fatal in childhood and adolescence, but some individuals live into adulthood.
Goltz Syndrome
Goltz syndrome, also known as focal dermal hypoplasia (FDH) or Goltz-Gorlin syndrome, is an X-linked dominant multisystem disorder that mainly affects the development of the skin, skeleton, eyes, and face. It is a type of ectodermal dysplasia, which is a group of hereditary disorders that cause the hair, teeth, nails, and glands to develop and function abnormally. It is a rare condition, with fewer than 300 cases reported, most of whom are female. It is caused by abnormalities of the PORCN gene on the X chromosome, which usually occur in the embryo and are not inherited from a parent. Goltz syndrome can affect almost any organ in the body, and the severity of the condition can vary greatly between individuals depending on the number and nature of the abnormalities. Skin manifestations usually present as patchy skin hypoplasia (thinning or absence of skin) that follows the lines of Blaschko (a pattern of skin stripes formed during embryonic development), as well as fat nodules and fat herniation, which are subcutaneous fat protruding through defects in the dermis and forming soft, yellow-pink nodules on the skin surface. Goltz syndrome can also cause structural abnormalities in other parts of the body. Some patients may also have problems with the gastrointestinal, genitourinary, neurological, and cardiovascular systems. The diagnosis of Goltz syndrome is mainly based on clinical manifestations and family history and can also be confirmed by genetic testing for PORCN gene variants. There is currently no curative treatment for Goltz syndrome; only symptomatic and supportive treatment can be given according to the specific situation of the patient, such as surgical repair of skin defects or malformed parts, medication to control infection or bleeding, etc. Gene therapy is a potential treatment method, but there have been no clinical trials or reported effective cases for Goltz syndrome so far.
Cornelia de Lange Syndrome
Cornelia de Lange syndrome (CdLS) is a rare genetic disorder that affects the development of many parts of the body. People with CdLS have a spectrum of physical, cognitive, and medical challenges that vary from mild to severe. Some of the common features of CdLS include long and/or thick eyebrows, a short nose, a long and/or smooth philtrum, a thin upper lip, missing fingers or toes, and a congenital diaphragmatic hernia. Other possible features include developmental delay or intellectual disability, small size and height, microcephaly, hearing impairment, vision abnormalities, gastroesophageal reflux, gastrointestinal abnormalities, musculoskeletal problems, scoliosis, social anxiety, seizures, cleft palate, and feeding problems. CdLS is caused by changes in one of five genes that are involved in the regulation of gene expression: NIPBL, SMC1A, SMC3, HDAC8, and RAD21. However, in about 30% of cases, the genetic cause of CdLS remains unknown. CdLS can be inherited from a parent who has the same genetic change, or it can occur for the first time in a person due to a new genetic change. The inheritance pattern can be autosomal dominant (NIPBL, SMC2, or RAD21) or X-linked (SMC1A or HDAC8). There is no specific treatment or cure for CdLS. The management of CdLS depends on the individual needs and symptoms of each person. It may involve nutritional support, physical therapy, speech therapy, occupational therapy, educational intervention, psychological and social support, medication, and surgery. Gene therapy is not currently available for CdLS, but some research studies are investigating its potential for other genetic disorders that affect the nervous system's development.
Alport Syndrome
Alport syndrome is a genetic disorder that affects the kidneys, ears, and eyes. It is caused by mutations in the genes encoding type IV collagen, a structural protein in the glomeruli, cochlea, and eyes. There are three genetic types of Alport syndrome: X-linked (XLAS), autosomal recessive (ARAS), and autosomal dominant (ADAS). The main symptoms of Alport syndrome are progressive kidney failure, leading to hematuria, proteinuria, hypertension, and edema. Additionally, patients may also have hearing loss and eye abnormalities such as cataracts, lens dislocation, retinal degeneration, and so on. The diagnosis of Alport syndrome relies on family history, clinical manifestations, urine tests, a kidney biopsy, and genetic testing.
There is no cure for Alport syndrome; only treatments to slow down or improve kidney damage exist, such as medication, dialysis, or kidney transplants. The common medication is to use angiotensin-converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB) to lower blood pressure and reduce proteinuria. Hearing loss can be improved by wearing hearing aids. Gene therapy has not yet achieved breakthroughs in the clinical application of Alport syndrome, but some laboratory studies are underway. For example, one study used a recombinant adeno-associated virus vector called AAV9-COL4A3 to deliver the normal COL4A3 gene into the glomerular cells of mice, thereby restoring the expression of type IV collagen and alleviating kidney injury. This study provides some hope for developing effective gene therapy strategies in the future.
References
- Islam YFK, et al. Incontinentia pigmenti and the eye. Curr Opin Ophthalmol. 2022 Nov 1;33(6):525-531.
- How KN, et al. Uncovering incontinentia pigmenti: From DNA sequence to pathophysiology. Front Pediatr. 2022 Sep 6;10:900606.
- Fukumoto S. FGF23-related hypophosphatemic rickets/osteomalacia: diagnosis and new treatment. J Mol Endocrinol. 2021 Feb;66(2):R57-R65.
- Baroncelli GI, et al. X-Linked Hypophosphatemic Rickets: Multisystemic Disorder in Children Requiring Multidisciplinary Management. Front Endocrinol (Lausanne). 2021 Aug 6;12:688309.
- Yamazaki M, et al. Osteocytes and the pathogenesis of hypophosphatemic rickets. Front Endocrinol (Lausanne). 2022 Sep 29;13:1005189.
- Crow YJ, et al. Treatments in Aicardi-Goutières syndrome. Dev Med Child Neurol. 2020 Jan;62(1):42-47.
- Tonduti D, et al. Novel and emerging treatments for Aicardi-Goutières syndrome. Expert Rev Clin Immunol. 2020 Feb;16(2):189-198.
- Spadari F, et al. Multidisciplinary approach to Gorlin-Goltz syndrome: from diagnosis to surgical treatment of jawbones. Maxillofac Plast Reconstr Surg. 2022 Jul 18;44(1):25.
- DiSalvo DS, et al. Pharyngeal Presentation of Goltz Syndrome: A Case Report with Review of the Literature. Head Neck Pathol. 2016 Jun;10(2):188-91.
- Kline AD, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018 Oct;19(10):649-666.
- Cornelia de Lange syndrome: from molecular diagnosis to therapeutic approach. J Med Genet. 2020 May;57(5):289-295.
- Selicorni A, et al. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes (Basel). 2021 Jul 15;12(7):1075.
- Kashtan CE. Alport Syndrome: Achieving Early Diagnosis and Treatment. Am J Kidney Dis. 2021 Feb;77(2):272-279.
- Warady BA, et al. Alport Syndrome Classification and Management. Kidney Med. 2020 Aug 7;2(5):639-649.
- RASTGAR, ASGHAR. "New concepts in pathogenesis of renal hypophosphatemic syndromes." (2009): 1-6.