Diagnosis of Rare Genetic Disorders
Diagnosis of Rare Genetic DisordersRare genetic disorders, such as Emanuel syndrome, Cat eye syndrome, Jacobsen syndrome, and Kleefstra syndrome, result from changes or mutations in the DNA sequence or structure of one or more genes and impact fewer than 1 in 2000 individuals in the general population.
These disorders present significant challenges in diagnosis and treatment. Firstly, these disorders are often difficult to identify due to their diverse and complex clinical manifestations, which can overlap with symptoms of more common diseases. Secondly, diagnosing rare genetic disorders may necessitate specialized genetic tests that are neither widely available nor affordable for all. Thirdly, many of these disorders lack effective therapies or cures, leaving healthcare providers to offer only palliative care or symptom management. Lastly, rare genetic disorders can carry psychosocial and ethical implications for patients and their families, including stigma, discrimination, social isolation, and challenging decision-making dilemmas. These factors complicate the overall management of individuals affected by these conditions.
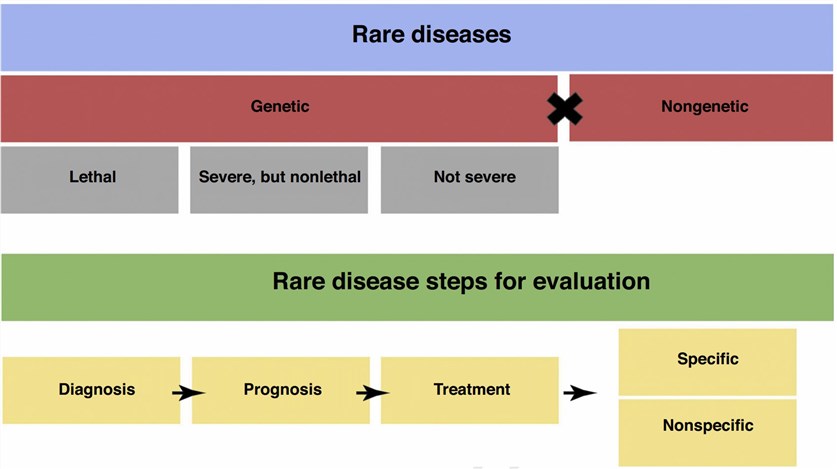 Fig.1 Rare disease stratification and steps for evaluation. (Pogue RE, 2018)
Fig.1 Rare disease stratification and steps for evaluation. (Pogue RE, 2018)
Emanuel Syndrome
Emanuel SyndromeEmanuel syndrome, a rare chromosomal disorder affecting approximately 1 in 110,000 live births, results from the fusion of parts of chromosome 11 and chromosome 22, forming an extra chromosome known as derivative 22 or der(22) chromosome. Individuals with Emanuel syndrome exhibit multiple congenital anomalies and developmental delays, including distinctive facial features, heart defects, kidney abnormalities, genital anomalies, skeletal irregularities, and neurological issues.
Diagnosis of Emanuel syndrome relies on clinical features and chromosomal analysis, which can detect the der(22) chromosome through various techniques. Molecular testing can further confirm the specific genes involved in the fusion.
The treatment approach for Emanuel syndrome is supportive and symptomatic, as there is no cure for this disorder. Therapeutic interventions may include surgical procedures, medications, and participation in early intervention programs. The prognosis of Emanuel syndrome varies based on the severity and type of anomalies present, as well as the potential complications that may arise from these abnormalities.
Cat Eye Syndrome
Cat Eye SyndromeCat eye syndrome, a rare chromosomal disorder occurring in approximately 1 in 50,000 to 1 in 150,000 live births, is characterized by an additional piece of chromosome 22 that is both inverted and duplicated. This extra chromosomal segment is referred to as an isodicentric chromosome 22 or idic(22) chromosome. Individuals with Cat Eye Syndrome exhibit multiple congenital anomalies and developmental delays, including distinctive eye abnormalities that resemble cat-like eyes, ear anomalies, anal atresia, kidney irregularities, heart defects, and skeletal abnormalities. The severity and specific features can vary among individuals, contingent upon the size and location of the idic(22) chromosome.
Diagnosis of Cat Eye Syndrome relies on clinical features and chromosomal analysis, utilizing various techniques to detect the idic(22) chromosome. Molecular testing can further confirm the specific genes involved in the duplication and inversion of chromosome 22.
The treatment approach for Cat Eye Syndrome is supportive and symptomatic, as there is currently no cure for this disorder. Interventions may include surgical procedures, medications, and participation in early intervention programs. The prognosis of Cat Eye Syndrome is determined by the severity and type of anomalies present, along with potential complications that may arise from these abnormalities.
Uniparental Disomy
Uniparental disomy (UPD) is a rare genetic phenomenon occurring when an individual inherits both copies of a chromosome or a segment from the same parent, instead of one copy from each parent. UPD can affect any chromosome or any part of a chromosome and is classified into two types: heterodisomy and isodisomy. Various mechanisms such as nondisjunction, anaphase lag, fertilization, or mitotic errors can lead to UPD by rescuing abnormal gametes or zygotes. UPD can cause a range of genetic disorders or phenotypic abnormalities, including Prader-Willi syndrome, Angelman syndrome, Beckwith-Wiedemann syndrome, Silver-Russell syndrome, and cystic fibrosis. Additionally, UPD can influence the expression of imprinted genes, which have distinct expression patterns based on parental origin and play vital roles in growth, development, metabolism, behavior, and cancer. The detection of UPD involves various techniques analyzing the chromosomal composition and the parental origin of the chromosomes. Examples of these techniques include karyotyping, FISH, microsatellite analysis, SNP array, and MSPCR. Molecular testing can also confirm the diagnosis of UPD by identifying the specific genes involved in the phenomenon and their imprinting status..
Jacobsen Syndrome
Jacobsen syndrome is a rare genetic disorder that affects about 1 in 100,000 live births. It is caused by a deletion at the end of the long arm of chromosome 11. The size and location of the deletion vary from person to person, often involving the loss of several genes crucial for the development and function of various organs and systems. Individuals with Jacobsen syndrome exhibit multiple congenital anomalies and developmental delays, including distinctive facial features, bleeding disorders, heart defects, immune system problems, skeletal anomalies, and neurological issues. Diagnosis of Jacobsen syndrome relies on clinical features and chromosomal analysis, which can detect the deletion of chromosome 11 through various techniques. Molecular testing can further confirm the specific genes affected by the deletion and their functions. Treatment for Jacobsen syndrome is primarily supportive and symptomatic, involving surgery, medication, and early intervention programs to manage the condition. Unfortunately, there is no cure for this disorder. The prognosis of Jacobsen syndrome depends on the severity and type of anomalies and the complications that may arise from them.
Kleefstra Syndrome
Kleefstra syndrome is a rare genetic disorder that affects about 1 in 100,000 live births. It is caused by a deletion or a mutation of the EHMT1 gene on chromosome 9. The EHMT1 gene encodes a protein that regulates gene expression and chromatin structure. Individuals with Kleefstra syndrome exhibit multiple congenital anomalies and developmental delays, including distinctive facial features, hypotonia, heart defects, urogenital anomalies, skeletal anomalies, and neurological problems. Diagnosis of Kleefstra syndrome relies on clinical features and genetic testing, which can detect the deletion or mutation of the EHMT1 gene using various techniques. Molecular testing can further identify the type and location of the deletion or mutation and their effects on protein function. Treatment for Kleefstra syndrome is primarily supportive and symptomatic, involving surgery, medication, and early intervention programs to manage the condition. Unfortunately, there is no cure for this disorder. The prognosis of Kleefstra syndrome depends on the severity and type of anomalies and the complications that may arise from them.
References
- Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet. 2010 Nov;128(5):441-52.
- Pogue RE, et al. Rare genetic diseases: update on diagnosis, treatment and online resources. Drug Discov Today. 2018 Jan;23(1):187-195.
- Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014 Jun 19;370(25):2418-25.
- Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013 May;14(5):307-20.
- Boerwinkle E, et al. A vision for the future of genomics research. Nature. 2003 Apr 24;422(6934):835-47.
- Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013 Feb;83(2):101-10.
- Cao H, Hegele RA. Identification of a novel mutation in the LPL gene in a patient with familial chylomicronemia syndrome by targeted next generation sequencing. J Clin Lipidol. 2016 May-Jun;10(3):687-92.
- Chong JX, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015 Aug 6;97(2):199-215.
- D’haene B, et al. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010 Apr;50(4):262-70.
- Gahl WA, et al. The NIH Undiagnosed Diseases Program and Network: applications to modern medicine. Mol Genet Metab. 2016 Feb;117(2):111-21.
- Gargis AS, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012 Nov;30(11):1033-6.
- Gilissen C, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014 Jul 17;511(7509):344-7.
- Hickey SE, et al. ACMG Practice Guideline: lack of evidence for MTHFR polymorphism testing. Genet Med. 2013 Feb;15(2):153-6.
- Lee H, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014 Nov 12;312(18):1880-7.
- Liu P, et al. Mechanism, prevalence, and more severe neuropathy phenotype of the Charcot-Marie-Tooth type 1A triplication. Am J Hum Genet. 2014 Feb 6;94(2):462-9.
- Lupski JR, et al. Clan genomics and the complex architecture of human disease. Cell. 2011 Sep 30;147(1):32-43.
- MacArthur DG, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014 Apr 24;508(7497):469-76.
- Neri G, et al. Clinical and molecular aspects of the Simpson-Golabi-Behmel syndrome. Am J Med Genet C Semin Med Genet. 2006 May 15;142C (2):140–52.