Uniparental Disomy
Uniparental disomy (UPD) is a rare genetic condition in which a person inherits both copies of a chromosome or a segment of a chromosome from one parent, instead of one copy from each parent. UPD can result from various mechanisms during meiosis or mitosis, and can cause various genetic diseases, depending on the type and location of the affected chromosome. UPD can affect autosomal chromosomes, sex chromosomes, and imprinted genes. Genomic imprinting is a process in which some genes are expressed differently depending on whether they are inherited from the father or the mother. The diagnosis of UPD is important for the accurate identification and management of genetic diseases, as well as for the genetic counseling and risk assessment of the affected families. However, the diagnosis of UPD is challenging due to the rarity and complexity of the condition, and the lack of standardized and comprehensive diagnostic methods.
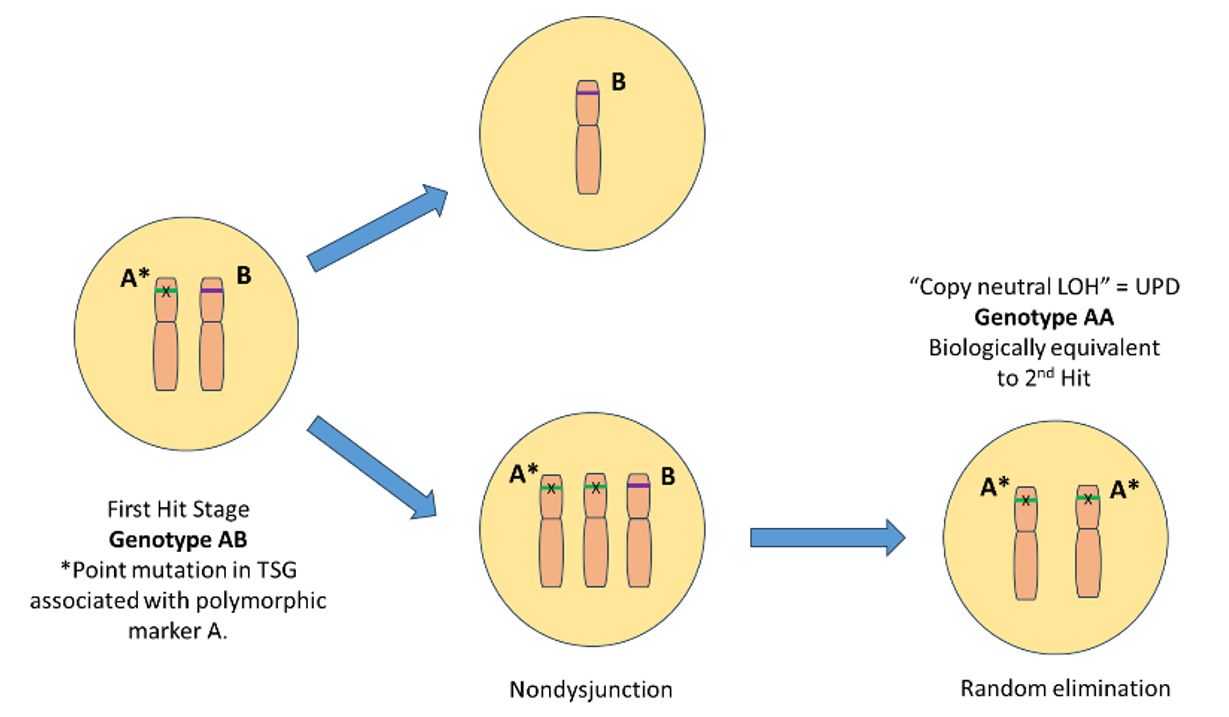 Fig.1 Uniparental Disomy (Creative Biolabs)
Fig.1 Uniparental Disomy (Creative Biolabs)
Clinical Manifestations
UPD can cause various genetic diseases with different clinical manifestations, depending on the type and location of the affected chromosome. Some of the most common genetic diseases caused by UPD are Prader-Willi syndrome (PWS), Angelman syndrome (AS), Beckwith-Wiedemann syndrome (BWS), Silver-Russell syndrome (SRS), and congenital hyperinsulinism (CHI). These diseases are associated with abnormal expression of imprinted genes on chromosomes 15, 11, or 7. Imprinted genes are genes that are expressed differently depending on whether they are inherited from the father or the mother. The clinical manifestations of these diseases are heterogeneous and variable, depending on several factors, such as the mechanism of UPD (heterodisomy or isodisomy), the size and location of the chromosomal abnormality, the presence or absence of uniparental heterodisomy, the type and extent of the imprinting defect (loss of methylation or gain of methylation), and the involvement of other genes or chromosomal regions. Moreover, environmental factors, such as nutrition, infection, stress, and medication, can also influence the expression and function of imprinted genes and affect the clinical outcome.
Clinical Diagnosis and Treatment Methods
UPD can be diagnosed by various methods that detect the presence or absence of parental alleles, haplotypes, chromosomes, or segments. These methods include molecular genetics, cytogenetics, and genomics. Molecular genetics methods use PCR, STR analysis, SNP genotyping, and other techniques. Cytogenetics methods use karyotype analysis, FISH, and other techniques. Genomics methods use microarray, WGS, and other techniques. These methods have different advantages and disadvantages in terms of sensitivity, specificity, accuracy, cost-effectiveness, and availability. For example, molecular genetics methods are highly sensitive and specific for UPD, but they require prior knowledge of the candidate genes or regions. Genomics methods are powerful for detecting small CNVs or UPiD, but they have high cost and complexity.
UPD can be treated by various methods that modulate the expression or function of the affected genes or pathways, or replace the diseased cells or tissues. These methods include pharmacological therapy, gene therapy, stem cell therapy, and others. Pharmacological therapy uses drugs to modulate the expression or function of the affected genes or pathways. Gene therapy uses viral vectors or gene editing tools to correct the defective genes or restore the normal expression. Stem cell therapy uses hematopoietic stem cells or induced pluripotent stem cells to replace the diseased cells or tissues. These methods have different feasibility and effectiveness, as well as potential risks and limitations. For example, gene therapy is promising and potentially curative, but it may have ethical issues or immune reactions. Stem cell therapy is innovative and regenerative, but it may have technical challenges or tumorigenicity.
Conclusion
UPD is a rare genetic condition in which a person inherits both copies of a chromosome or a segment of a chromosome from one parent. UPD can cause various genetic diseases, depending on the type and location of the affected chromosome. UPD can affect autosomal chromosomes, sex chromosomes, and imprinted genes. The clinical manifestations, diagnosis, and treatment of UPD are heterogeneous, variable, and challenging, depending on several factors, such as the mechanism of UPD, the size and location of the chromosomal abnormality, the type and extent of the imprinting defect, and the environmental factors. UPD is a fascinating and important topic in human genetics, but there are still many challenges and uncertainties in the field of UPD research. More studies are needed to improve our understanding and management of this rare genetic condition.
References
- Martin AR, et al. Population structure and the effects of ancestry on uniparental disomy. Nat Commun. 2020 Jan 10;11(1):219.
- Kotzot D, et al. Uniparental disomy 16: two new cases and a review of the literature. Prenat Diagn. 2005 May;25(5):362-8.
- Zhang X, et al. Case Report: Paternal Uniparental Isodisomy and Heterodisomy of Chromosome 16 With a Normal Phenotype. Front Pediatr. 2021 Oct 22;9:732645.
- Liehr T. Uniparental disomy is a chromosomic disorder in the first place. Mol Cytogenet. 2022 Feb 17;15(1):5.
- Eggermann T, et al. Uniparental disomy (UPD) syndromes: clinical and molecular aspects. J Mol Med (Berl). 2004 Mar;82(3):161-8.
- Eggermann T, et al. Complex and segmental uniparental disomy (UPD): review and lessons from rare chromosomal complements. J Med Genet. 2001 Aug;38(8):497-507.
- Kotzot D, et al. Maternal uniparental disomy 7 and Silver-Russell syndrome - clinical update and comparison with other subgroups. Eur J Hum Genet. 2015 Sep;23(9):1101-8.
- Buiting K, et al. Clinical features of maternal uniparental disomy 14 in patients with an epimutation and a deletion of the imprinted DLK1/GTL2 gene cluster. Hum Mutat. 2008 Jul;29(7):1141-6.
- Eggermann T, et al. Congenital hyperinsulinism: molecular basis of uniparental disomy of chromosome 11p15 and its impact on genetic counseling and counseling for recurrence risk. Mol Syndromol. 2014 Jan;5(1):10-6.
- Buiting K, et al. Molecular diagnosis and clinical management of Prader-Willi syndrome: practical recommendations of the German/Austrian/Swiss expert panel. Mol Syndromol. 2018 Feb;9(1):28-37.
- Williams CA, et al. Angelman syndrome: consensus for diagnostic criteria. Am J Med Genet A. 2006 Mar 15;140(6):413-8.
- Brioude F, et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018 Apr;14(4):229-49.