


Whole Exome Sequencing (WES) based Bioinformatic Analysis Service
WES Services Features Q&As Resource
Whole Exome Sequencing (WES)
- Overview
Whole Exome Sequencing (WES) is an incredibly potent genomic technique. It zeroes in on sequencing the exonic regions—the portions of the genome that directly encode proteins. Now, while exons make up only 1-2% of the entire human genome, they pack a punch: these small segments contain roughly 85% of all known disease-causing mutations. This astonishing concentration of critical information makes WES a go-to method for pinpointing genetic variants that may be linked to inherited disorders or diseases that have a strong genetic basis. It's a remarkably efficient, and comparatively cost-effective, approach.
The way WES works is by selectively capturing and sequencing the exome, the coding regions of the genome, while leaving out non-coding regions such as introns and intergenic sequences. With the advent of high-throughput sequencing technologies, WES has the capability to churn out massive amounts of data. This data can then be meticulously analyzed to uncover single nucleotide variants (SNVs), small insertions and deletions (indels), and other genetic alterations. Its precision makes it particularly powerful in studying rare Mendelian disorders, where the root cause may lie in a mutation of a single gene. These are diseases where every genetic clue counts.
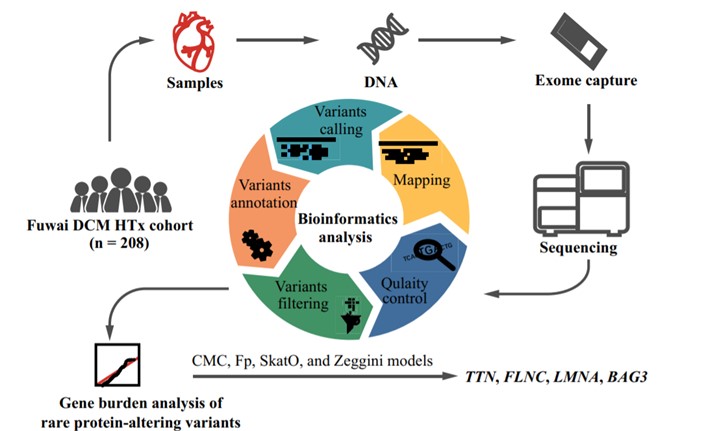 Fig.1 WES workflow.1
Fig.1 WES workflow.1
But that's not all—WES has profound applications in cancer research. Within tumor cells, WES is deployed to uncover somatic mutations, those non-inherited changes that occur in cells as cancer develops. These mutations often play a role in cancer progression, and pinpointing them is vital. Understanding which genes have gone awry and are fueling a tumor's growth opens the door to developing highly targeted therapies. This approach is revolutionizing cancer treatment, tailoring therapies to attack the genetic vulnerabilities specific to each patient's tumor.
And there's more—WES is also leaving its mark in pharmacogenomics. As precision medicine continues to advance, WES helps shed light on how individual genetic variations influence a person's response to medications. This means that by understanding the intricacies of a patient's exome, clinicians can create treatment plans that are not just effective but highly personalized, designed with the individual patient's genetic profile in mind. Personalized medicine isn't just on the horizon; it's already making headway, thanks in large part to WES.
- WES Workflow
WES for cancer involves the following key steps
- Sample Collection and Preparation: Tumor and matched normal tissue samples are collected.
- DNA Extraction: Genomic DNA is isolated from the samples.
- Library Preparation: DNA is fragmented, adapters are added, and the library is amplified.
- Exome Capture: Exonic regions of the genome are selectively enriched using probes.
- Next-Generation Sequencing (NGS): The enriched libraries undergo sequencing through high-throughput technologies.
- Data Analysis: Data filtration, alignment, basic analysis, advanced analysis, and customized analysis.
Cancer WES Bioinformatic Analysis at Creative Biolabs
- Importance
WES data analysis is vital for extracting meaningful insights from sequencing results, especially in cancer research. It enables the identification of disease-causing mutations by focusing on protein-coding regions, where most pathogenic mutations occur. In cancer, WES data analysis is crucial for detecting somatic mutations that drive tumor growth, helping to distinguish these from inherited germline variants. This distinction is essential for understanding tumor biology and identifying actionable mutations that can inform targeted therapies.
Data analysis also plays a critical role in annotating and interpreting the functional impact of identified variants, which is necessary for prioritizing those with clinical significance. Furthermore, the analysis involves rigorous quality control to ensure the accuracy and reliability of results. In cancer sequencing, WES data analysis guides personalized medicine by linking specific genetic alterations to potential treatment options, ultimately improving patient outcomes. The ability to filter, prioritize, and interpret vast amounts of sequencing data makes WES analysis indispensable in cancer diagnostics and therapeutic decision-making.
- Workflow
Creative Biolabs has developed a powerful in-house toolkit for cancer WES bioinformatic analysis with highly accurate interpretation. The workflow of our general WES analysis is as follows:
-
 Quality
Quality
control of
raw data -
Mapping
of the
clean data -
Production
of the
clean data -
Post-alignment processing
-
Quality control of the mapping reads
-
 Variant calling, annotation and prioritization
Variant calling, annotation and prioritization
- Key Notes for WES Bioinformatic Analysis
The following section outlines key considerations and best practices for ensuring accuracy and reliability in WES bioinformatic analysis:
1. Quality Control of Sequencing Data
Ensuring high-quality data is essential. After sequencing, reads should be assessed for quality using tools like FastQC to remove low-quality reads, trim adapters, and filter out sequencing artifacts. This step is crucial for minimizing errors in downstream analysis.
2. Accurate Read Alignment
Reads must be properly aligned to a reference genome using alignment tools like BWA or Bowtie2. Misalignments can lead to incorrect variant calls, so this step requires careful validation to ensure reads are correctly mapped to the exonic regions of the genome.
3. Variant Calling
Accurate identification of variants (e.g., SNVs, indels) is critical. The variant calling process requires specialized tools (e.g., GATK, Mutect2) to distinguish true variants from sequencing noise. For cancer, identifying somatic mutations by comparing tumor and normal samples is crucial, and germline mutations should be filtered out.
4. Variant Annotation
Once variants are identified, they must be annotated using tools like ANNOVAR or VEP to assess their functional impact. Annotation helps determine whether a variant affects gene function, is linked to known diseases, or may be clinically relevant. Annotation tools also provide information on whether the mutation is known in databases like COSMIC or ClinVar.
5. Prioritization and Filtering
Given the large number of variants, prioritization is essential. Filters are applied to focus on variants with high pathogenic potential, such as those with low population frequency, those found in cancer-related genes, or those affecting protein-coding regions. This helps narrow down the results to clinically or biologically significant mutations.
Key Features and Advantages of Cancer WES Bioinformatics Analysis
- Highly accurate interpretation.
- Rapid and comprehensive discovery.
- Excellent sensitivity for detecting copy number variations (CNVs).
- Improved coverage of challenging genes.
- Secured and long-term secure cloud storage.
Equipped with world-leading technology platforms and professional scientific staff, Creative Biolabs has accumulated extensive experience in cancer WES and data analysis. Through our high-throughput and automated system, clients could accelerate their research with the most comprehensive information in a time-saving manner. We are pleased to use our extensive experience and advanced platform to offer the best service to satisfy each unique demand from our clients.
Please contact us for more information and a detailed quote.
Q&As
Q: What is the role of functional annotation in WES Bioinformatics Analysis?
A: Functional annotation links genetic variants to biological functions and pathways. In WES Bioinformatics Analysis, this process helps researchers understand the impact of identified mutations on protein function and disease mechanisms, aiding in the prioritization of variants for further study.
Q: What are the main features of WES Bioinformatics Analysis?
A: Key features include the identification of SNVs, indels, CNVs, and functional annotations. These features provide a comprehensive view of genetic mutations affecting protein-coding regions.
Q: What is the significance of sequencing depth in WES?
A: Sequencing depth, typically recommended at 100X for WES, ensures robust detection of genetic variants, including rare mutations. Higher depth increases the confidence in variant calls, making the analysis more accurate and reliable.
Q: What bioinformatics tools are used in WES analysis?
A: Common bioinformatics tools used in WES analysis include sequence alignment software, variant calling algorithms, and annotation tools. These tools help process and interpret exome sequencing data, enabling the identification of genetic mutations and their potential impacts.
Q: How can WES be applied to cancer research?
A: WES can identify mutations in the exome that drive cancer progression. By analyzing these mutations, researchers can understand the genetic basis of cancer, discover new therapeutic targets, and develop personalized treatment strategies based on the genetic profile of the tumor.
Reference
- Lian, Hong, et al. "Genetic characterization of dilated cardiomyopathy patients undergoing heart transplantation in the Chinese population by whole-exome sequencing." Journal of Translational Medicine 21.1 (2023): 476. Distributed under Open Access license CC BY 4.0, without modification.
Resources
Infographics
Podcast
Note: Our sequencing services are for Research Use Only. Not For Clinical Diagnosis.
Related Services:
- WGS Bioinformatic Analysis
- Targeted Sequencing Bioinformatic Analysis
- WTS Bioinformatic Analysis
- SuPrecision™ Pipeline
Online Inquiry
