Cystic Fibrosis
Cystic fibrosis (CF) is a genetic disorder that affects the respiratory and digestive systems, leading to abnormal mucus secretion and infections. It stands as one of the most common life-threatening inherited diseases, impacting approximately 1 in 2,500 newborns in Europe and North America. CF arises from mutations in the CFTR gene, responsible for encoding a protein regulating chloride ion transport across cell membranes. Defective CFTR protein functionality compromises various organs, especially the lungs and pancreas. CF follows an autosomal recessive inheritance pattern, requiring mutations in both copies of the CFTR gene for a person to have CF.
Clinical Manifestations of Cystic Fibrosis
Symptoms of CF vary depending on patients' age and organ involvement. A persistent cough with thick, difficult-to-clear mucus is a common and defining symptom, resulting in chronic lung infections, inflammation, and damage. Respiratory symptoms also encompass wheezing, shortness of breath, chest pain, and nasal polyps. CF affects the digestive system, leading to nutrient malabsorption, weight loss, failure to thrive, and pancreatic insufficiency. Patients may develop diabetes, liver disease, gallstones, or intestinal obstruction. Infertility is common in males and may reduce fertility in some females due to abnormal reproductive organ development or obstruction. The severity and progression of CF vary widely among individuals, influenced by the specific mutations in the CFTR gene.
Diagnosis and Treatment of Cystic Fibrosis
CF diagnosis relies on clinical criteria and laboratory tests. Clinical criteria include typical symptoms, family history of CF, or a positive newborn screening test. Laboratory tests measure elevated chloride concentration in sweat, a common trait in CF patients, and conduct genetic testing to identify CFTR gene mutations. Couples at risk of having a child with CF can opt for prenatal screening and diagnosis. CF treatment aims to enhance patients' quality and length of life by managing symptoms and complications.
Treatment options encompass pharmacological therapy, incorporating antibiotics, anti-inflammatory drugs, mucolytics, and CFTR modulators targeting the underlying CFTR protein defect. Physical therapy, including chest physiotherapy and exercise, aids in clearing mucus from the lungs and improving lung function. Nutritional support, such as pancreatic enzyme supplements and a high-calorie diet, is crucial to prevent malnutrition and growth failure. For patients with end-stage lung disease meeting eligibility criteria, lung transplantation may be considered.
Conclusion
CF is a complex and diverse genetic disorder affecting multiple organs and systems. Its diagnosis combines clinical and laboratory criteria, with treatment involving a multidisciplinary approach addressing various disease aspects. Despite advances in understanding and managing CF, challenges persist, including enhancing CFTR modulator accessibility and affordability, developing gene therapy or gene editing strategies, improving early detection and prevention, and reducing disparities in CF care. Promising future research and treatment avenues, including emerging technologies and therapies undergoing clinical trials, aim to ultimately cure CF and enhance patients' and their families' lives.
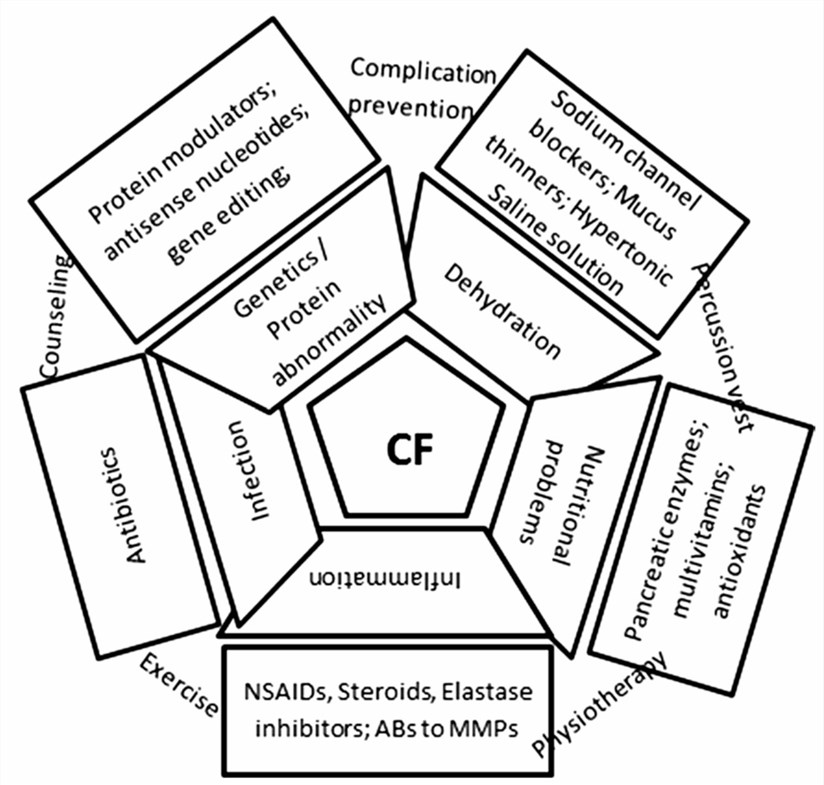 Fig.1 The main pathophysiological dysfunctions and treatment modalities for CF patients.
Fig.1 The main pathophysiological dysfunctions and treatment modalities for CF patients.
References
- Allan KM, et al. Treatment of Cystic Fibrosis: From Gene- to Cell-Based Therapies. Front Pharmacol. 2021 Mar 16;12:639475.
- Brody H. Cystic fibrosis. Nature. 2020 Jul 29;583:S1.
- Cutting GR. Cystic Fibrosis: A Review. JAMA. 2021 Sep 14;326(10):947-956.
- Davies JC, et al. Cystic fibrosis. Lancet. 2007 Jan 27;369(9559):390-401.
- De Boeck K, et al. Cystic fibrosis: the end of an era? Eur Respir J. 2019 Dec 19;54(6):1901690.
- Elborn JS. Cystic fibrosis. Lancet. 2016 Nov 19;388(10059):2519-2531.
- Flume PA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007 Nov 15;176(10):957-969.
- Gibson RL, et al. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003 Oct 15;168(8):918-951.
- Griesenbach U, et al. Gene therapy for cystic fibrosis lung disease: current status and future perspectives. Curr Opin Pulm Med. 2013 Nov;19(6):651-657.
- Li W, et al. Advances in diagnosis and treatment of cystic fibrosis: a meeting report from the 17th International Conference on Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators in London, UK, October 2019. J Thorac Dis. 2020 Feb;12(2):106-113.
- Milla CE, et al. Nutritional status as a predictor of survival in patients with cystic fibrosis. Chest. 1996 Nov;110(5):1197-1202.
- O’Sullivan BP, et al. Cystic fibrosis. Lancet. 2009 May 30;373(9678):1891-1904.
- Ramsey BW, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011 Nov 3;365(18):1663-1672.
- Rafeeq MM, Murad HAS. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017 Apr 27;15(1):84.