Tay Sachs Disease
Tay Sachs disease is a rare and fatal inherited disorder of the nervous system that causes progressive deterioration of nerve cells and their functions. It is caused by mutations in the HEXA gene, which encodes the alpha subunit of the enzyme beta-hexosaminidase A. This enzyme is responsible for breaking down a fatty substance called GM2 ganglioside in the lysosomes of cells. When the enzyme is deficient or defective, GM2 ganglioside accumulates in the lysosomes, especially in the nerve cells of the brain and spinal cord, leading to neuronal damage and death. Tay Sachs disease affects people of all ethnic backgrounds, but it is more common among individuals of Ashkenazi Jewish, French Canadian, and Cajun descent. Among Ashkenazi Jews, the incidence of Tay Sachs disease is about 1 in 3,000 live births, compared to 1 in 320,000 live births in the general population.
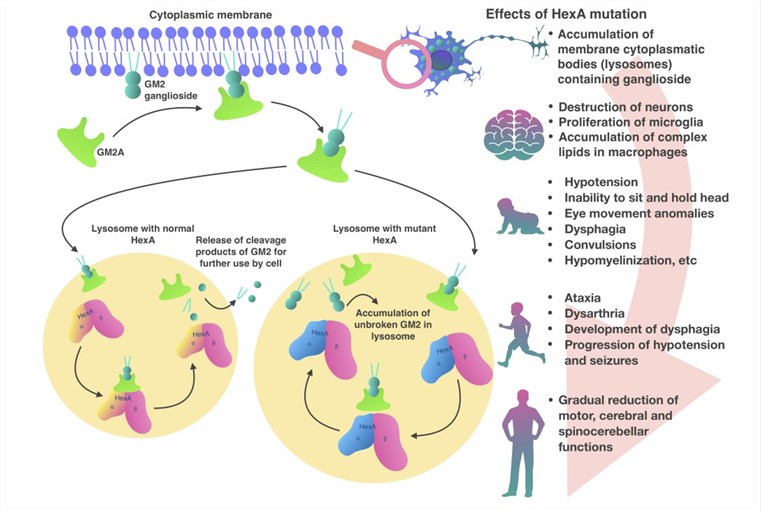 Fig.1 Pathogenesis of Tay-Sachs disease. (Solovyeva VV, 2018)
Fig.1 Pathogenesis of Tay-Sachs disease. (Solovyeva VV, 2018)
Clinical Manifestations
The clinical manifestations of Tay Sachs disease vary depending on the age of onset and the severity of the condition. There are three main forms of Tay Sachs disease: infantile, juvenile, and adult.
The infantile form is the most common and severe form of Tay Sachs disease. It usually appears in the first 6 months of life and is characterized by a rapid decline in mental and physical abilities. Infants with Tay Sachs disease typically develop normally until 3 to 6 months of age, after which they start showing signs of developmental delay, such as loss of motor skills, decreased eye contact, and an increased startle response. As the disease progresses, they develop other symptoms, including seizures, blindness, deafness, spasticity, cherry-red spots on the retina, and a characteristic "doll-like" facial appearance. Most children with infantile Tay Sachs disease do not survive past the age of 4.
The juvenile form is a less common and less severe variant of Tay Sachs disease. It usually manifests between 2 and 10 years of age, and is characterized by a slower decline in mental and physical abilities. Children with juvenile Tay Sachs disease may have a period of normal development before showing signs of cognitive impairment, such as learning difficulties, memory loss, and behavioral problems. They may also exhibit other symptoms such as ataxia, dysarthria, dysphagia, muscle weakness, and vision loss. Life expectancy for children with juvenile Tay Sachs disease varies, but most do not survive beyond their late teens or early twenties.
The adult form is the rarest and mildest variant of Tay Sachs disease. It usually emerges in adulthood and is marked by a gradual onset of neurological symptoms. Adults with Tay Sachs disease may have had a normal development until they start exhibiting signs of psychiatric disorders, such as depression, anxiety, psychosis, or bipolar disorder. Other symptoms in adults may include tremors, dystonia, chorea, or neuropathy. The progression and prognosis for adults with Tay Sachs disease vary, but most have a normal lifespan.
Diagnosis and Treatment Methods
The diagnosis and treatment of Tay Sachs disease are challenging and require a multidisciplinary approach. The diagnosis relies on a combination of clinical features, biochemical tests, and genetic tests. Biochemical tests measure the activity of beta-hexosaminidase A in blood, urine, or tissue samples. Low or absent enzyme activity indicates a potential Tay Sachs diagnosis. However, biochemical tests alone are insufficient for confirmation, as other conditions can cause similar enzyme deficiencies, such as Sandhoff disease or GM2 activator deficiency. Therefore, genetic tests are essential to identify specific mutations in the HEXA gene responsible for Tay Sachs disease. Genetic tests are also used for prenatal diagnosis and carrier screening, crucial for preventing and managing the disease in high-risk populations.
Treatment for Tay Sachs disease is primarily supportive and palliative, as there is no cure for the condition. The treatment aims to alleviate symptoms, improve patients' quality of life, and support their families. It may include medications to manage seizures, pain, and infections; physical therapy to maintain muscle tone and mobility; occupational therapy to assist with daily activities; speech therapy to enhance communication; nutritional support to prevent malnutrition and dehydration; and psychological counseling to help cope with emotional stress. Several experimental therapies, such as enzyme replacement therapy, gene therapy, stem cell therapy, and substrate reduction therapy, are under development. These therapies aim to restore beta-hexosaminidase A function or reduce GM2 ganglioside accumulation. However, these treatments are still in early stages of research and have not been proven safe and effective for human use.
Conclusion
Tay Sachs disease is a devastating genetic disorder that severely impacts the nervous system, causing significant disability and often resulting in premature death. Despite advancements in diagnosis and treatment, there is currently no cure, and management is predominantly supportive and palliative. Urgent research is needed to explore innovative therapies that could prevent, delay, or reverse Tay Sachs disease's progression. Promising research directions include improving the delivery and targeting of enzyme replacement therapy and gene therapy, investigating stem cell therapy and substrate reduction therapy, identifying new biomarkers and drug targets, and understanding the molecular mechanisms and pathways involved in Tay Sachs disease. By raising awareness and fostering collaboration among researchers, clinicians, patients, and families, there is hope that a cure for Tay Sachs disease will be discovered in the future.
References
- Solovyeva VV, et al. New Approaches to Tay-Sachs Disease Therapy. Front Physiol. 2018 Nov 20;9:1663.
- Ferreira CR, Gahl WA. Lysosomal Storage Diseases. Transl Sci Rare Dis. 2017;2(1-2):1-71.
- Sandhoff K, Harzer K. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci. 2013 Jul 10;33(28):10195-208.
- Lew RM, et al. Tay-Sachs disease: current perspectives from Australia. Appl Clin Genet. 2015 Feb 23;8:55-66.
- Solovyeva VV, et al. New Approaches to Tay-Sachs Disease Therapy. Front Physiol. 2018 Nov 20;9:1663.
- Picache JA, et al. Therapeutic Strategies for Tay-Sachs Disease. Front Pharmacol. 2022 Jul 5;13:906647.
- Sakuraba H, et al. Molecular heterogeneity of hexosaminidase A deficiency: identification and expression of two novel alpha-subunit mutations (Q170H and R178C) causing Tay-Sachs disease among Japanese patients. Biochim Biophys Acta. 2006 Jan;1762(1):45-52.
- Mistri M, T et al. Identification of novel mutations in HEXA gene in children affected with Tay Sachs disease from India. PLoS One. 2012;7(9):e39122.
- Leal NS, et al. GM2-Gangliosidoses: From Pathophysiology to Treatment Strategies-An Update on the Current Status and Future Perspectives. Int J Mol Sci. 2020 Sep 17;21(18):6814.
- Frisch A, et al. Origin and spread of the 1278insTATC mutation causing Tay-Sachs disease in Ashkenazi Jews: genetic drift as a robust and parsimonious hypothesis. Hum Genet. 2004 Mar;114(4):366-76.
- Di Rocco M, et al. Miglustat for the treatment of lysosomal storage disorders: a systematic review and meta-analysis of randomized controlled trials. Orphanet J Rare Dis. 2020 Dec 7;15(1):342.
- Platt FM, et al. Lysosomal storage diseases. Nat Rev Dis Primers. 2018 Nov 15;4(1):27.
- Bembi B, et al. Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J Inherit Metab Dis. 2010 Dec;33(6):727-35.