With years of experience and high-end technologies, Creative Biolabs has successfully developed a series of innovative and diversified assay platforms to provide fast and convenient services for our customers. We offer high-quality genetic/epigenetic analysis services for customers all over the world.
Genetic and epigenetic analysis is an important aspect of stem cell research as it helps scientists better understand the genetic and epigenetic mechanisms that control stem cell behavior and differentiation. By examining the genetic and epigenetic profiles of stem cells, researchers can identify key genes and pathways that regulate stem cell self-renewal and differentiation.
Genetic analysis involves studying the DNA sequences of stem cells to identify genetic variations and mutations that may impact stem cell function. Epigenetic analysis, on the other hand, focuses on studying the chemical modifications of DNA that can influence gene expression without changing the underlying DNA sequence. Epigenetic changes play a crucial role in regulating stem cell differentiation and maintaining stem cell identity.
Massively parallel sequencing technology lays the foundation for the construction of epigenomics. Creative Biolabs provides key sequencing-based methods used in the analysis of epigenomes.
5-methyl-cytosine (5mC) and its oxidized derivatives are measured on a genome-wide basis using enrichment and transformation methods followed by massively parallel sequencing. Bisulfite conversion provides a quantitative measurement of 5mC but does not distinguish 5hmC. Antibody enrichment provides qualitative measurements of 5mC and 5hmC. The bisulfite converted or enriched DNA is purified, library constructed and clonally sequenced. A special algorithm is needed to align the bisulfite conversion readings with the reference genome.
The genomic location of the modified histones is measured genome-wide by chromatin immunoprecipitation followed by massively parallel sequencing (ChIP-seq). Histones can be released from the genome by ultrasound, enzymatically digested, or inserted through a transposon. If sonication is used, chromatin must first be chemically cross-linked. After histone release, specific chemical modifications are enriched by immunoabsorption. Purification of enriched histone-associated DNA, which can be purified for high-throughput sequencing, provides complete information on specific DNA-protein interactions and binding sites.
The genomic location of long-range chromatin exposure is measured genome-wide by large-scale parallel sequencing of DNA fragments generated by adjacent ligation. Complete chromatin cross-linking physically connects adjacent genomic distal nucleosomes in 3-dimensional space. The cross-linked chromatin is enzymatically digested, and the resulting DNA ends are labeled with biotin and ligated adjacently. The ligated DNA is sheared by sonication or enzymatic digestion and the linked ligation is enriched by streptavidin pull-down. The resulting DNA is purified, library constructed and clonally sequenced.
Large-scale parallel sequencing of DNA fragments released from intact chromatin by transposon insertion, enzymatic digestion or sonication can measure the location of open chromatin in the genome-wide range. The obtained DNA fragment is subjected to size selection or phenol-chloroform extraction to eliminate nucleosome-associated DNA. The resulting DNA is purified, library constructed and sequenced, and aligned to the reference genome. Sequencing requirements depend on experimental parameters and resolution requirements, and range from 10 s to 100 s millions of fragments per sample.
Our team of experienced scientists and bioinformaticians work closely with researchers to design customized experimental strategies, analyze complex genomic datasets, and interpret biological findings.
Overall, our genetic and epigenetic analysis services are tailored to meet the specific needs of stem cell researchers, providing a comprehensive understanding of the molecular mechanisms governing stem cell behavior.
With the help of our well-established technology and experienced scientists, in addition to providing quality technical services, we offer a very flexible choice for each specific epigenetic project design and implementation. Please don't hesitate to contact us for more information.
Below are the findings presented in the article related to the genetic/epigenetic assays for stem cells.
Bogdan Andrei Mirauta et al. performed the first comprehensive proteomic analysis of iPSC, analyzing 202 iPSC lines from 151 donors and integrating transcriptomic and genomic sequence data from the same lines. They reported the first in-depth characterization of the human iPSC proteome, linking genetic variation to changes at the RNA and protein levels. Their data provided matched quantitative proteomics (tandem mass spectrometry) and transcriptomics (RNA-Seq) mapping of 202 iPSC lines. They mapped more than 600 cis protein quantitative trait loci (pQTL) and analyzed how they relate to cis eQTL, how they affect other proteins in trans, and how pQTL are associated with human disease variation.
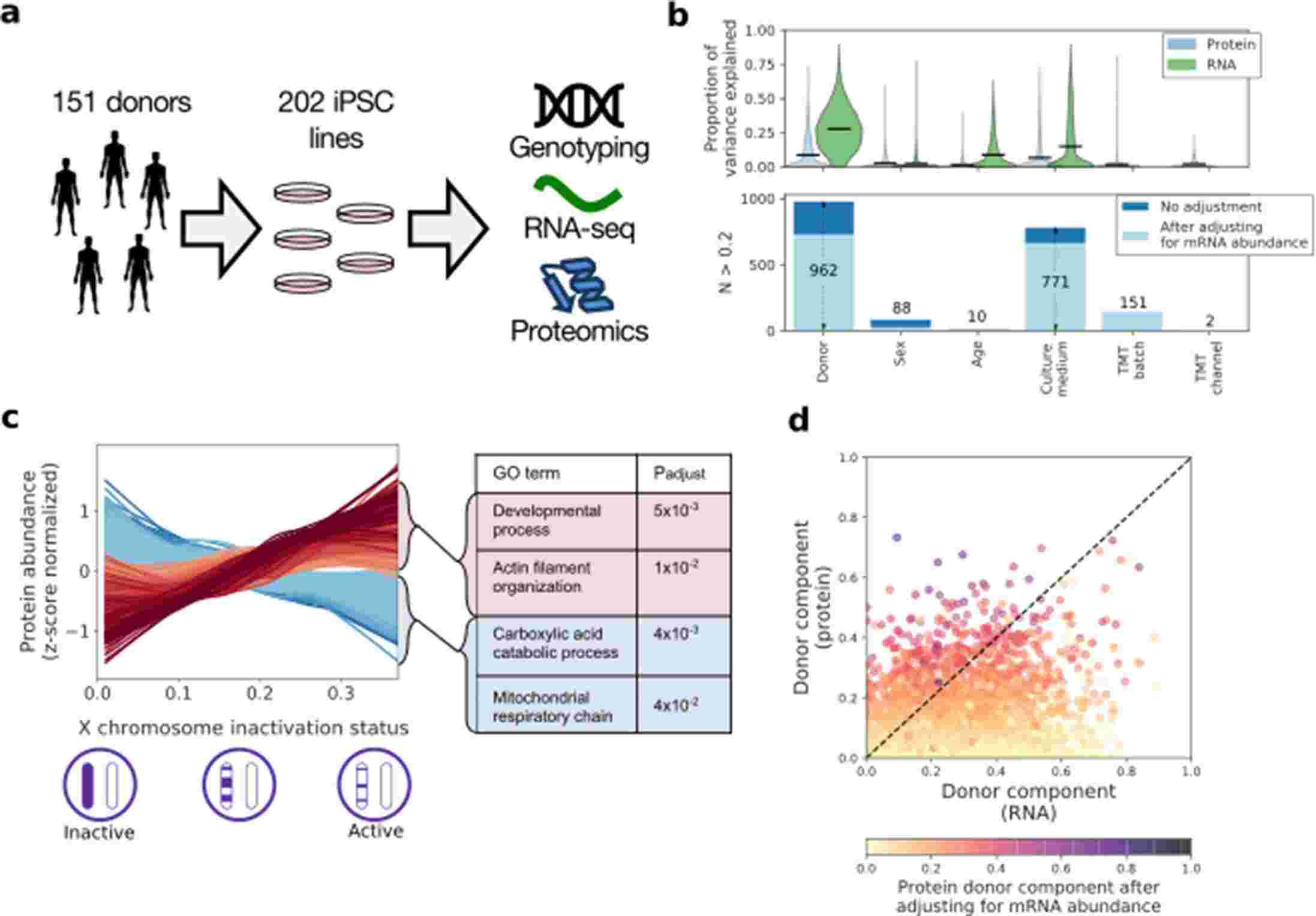 Fig. 1 Characterising variation in the iPSC proteome and transcriptome.1
Fig. 1 Characterising variation in the iPSC proteome and transcriptome.1
Reference
For Research Use Only. Not For Clinical Use.
