

NAA and Thalassemia
Thalassemias are inherited hematological diseases characterized by decreased hemoglobin production. As a genetic disorder, the relationship between thalassemia and natural autoantibody (NAA) has been revealed, which provides a novel approach to the diagnosis and treatments of thalassemia. Based on rich experience in antibody development and detection, Creative Biolabs has promoted continuous research and exploration in the field of NAA. Therefore, we can promise high-quality NAA services and high-performance NAA detection kits confidently to our clients all over the world.
Introduction of Thalassemia
Thalassemia is a genetic disorder that has been divided into two main categories including α-thalassemia and β-thalassemia, which involves abnormal haemoglobin formation. Haemoglobin comprises of α and β chains, which are faulty respectively in a patient with α or β thalassemia and result of which the haemoglobin produced is faulty. Therefore, 4 genes for α-globin or 2 genes for β-globin missing always result in various degrees of thalassemia. While thalassemia can be diagnosed via a complete blood count, hemoglobin electrophoresis or high-performance liquid chromatography, and DNA testing, the antenatal examination is still the most effective preventive measure.
Thalassemia Develop Pathway
We can set β-thalassemia as an example and summarize the pathway of thalassemia. During every stage of development, the production of α and β globin chains is balanced. However, in patients with β-thalassemia, impaired production of b-globin chains results in an excess of free α-globin chains that undergo denaturation and degradation in mature erythroid cells and their precursors to play a critical role in the causation of thalassemia. When the capacity of a hemoglobin stabilizing protein is exceeded, highly unstable free α-globin molecules undergo auto-oxidation, forming α-hemichromes and reactive oxygen species (ROS) to trigger cascades of events leading to hemolysis and ineffective erythropoiesis, which are the two primary pathophysiological mechanisms causing anemia in patients with β-thalassemia. ROS-induced alterations in membrane deformability and stability through partial oxidation of protein band 4.1 and defective assembly of spectrin-actin-band 4.1 membrane skeleton complex are believed to be the primary mechanism of hemolysis in mature red cells. Moreover, heat shock protein 70 interacts directly with free α-globin chains and thereby becomes sequestered in the cytoplasm. This, in turn, prevents the protein from performing its normal physiological role of protecting GATA-binding factor 1 (GATA1) from proteolytic cleavage. The resulting premature degradation of GATA1 results in maturation arrest and apoptosis of polychromatic erythroblasts.
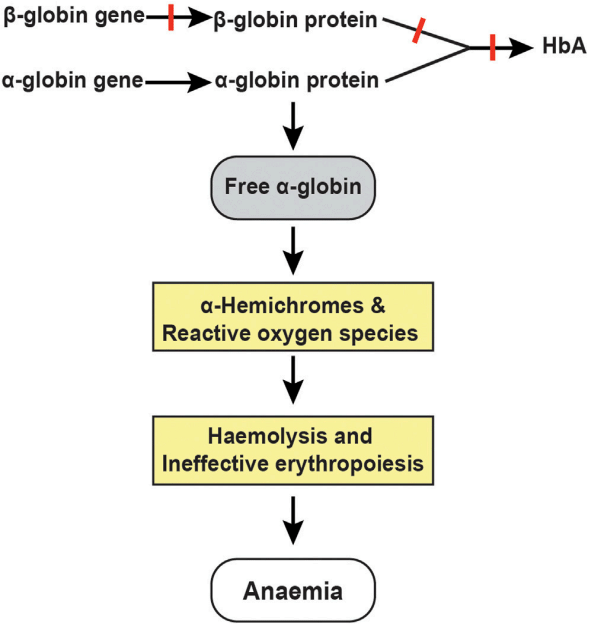 Fig.1 Clinical complications and pathophysiological mechanisms of β-thalassemia.1
Fig.1 Clinical complications and pathophysiological mechanisms of β-thalassemia.1
NAA Services for Thalassemia at Creative Biolabs
- NAA Services for Anti-ATP Synthase
- NAA Services for Anti-Alloantibody K
- NAA Services for Anti-Alloantibody E
- NAA Services for Anti-Alloantibody C
- NAA Services for Anti-Alloantibody JKb
- NAA Services for Anti-Alloantibody Cw
- NAA Services for Anti-Alloantibody D
- NAA Services for Anti-Alloantibody Xg
Thalassemia Related Products at Creative Biolabs
Creative Biolabs provides various NAA detection kits with high specificity and sensitivity which can act as a one-stop toolbox for studying disease pathways with NAA and screening for potential biomarkers.
Based on our established platforms and skilled employees, Creative Biolabs has achieved many remarkable goals in the NAA research and offered satisfactory services to global clients. Our high-quality products and services will contribute greatly to the success of your projects. Please feel free to contact us for more information and a detailed quote.
Reference
- Mettananda, Sachith. "Genetic and epigenetic therapies for β-thalassaemia by altering the expression of α-globin gene." Frontiers in Genome Editing 3 (2021): 752278.