Creative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsCreative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsCreative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsCreative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsWith over a decade of experience in phage display technology, Creative Biolabs can provide a series of antibody or peptide libraries that are available for licensing or direct screening. These ready-to-use libraries are invaluable resources for isolating target-specific binders for various research, diagnostic or therapeutic applications.
HighlightsCreative Biolabs has established a broad range of platforms for developing novel antibodies or equivalents. These cutting-edge technologies enable our scientists to meet your demands from different aspects and tailor the most appropriate solution that contributes to the success of your projects.
HighlightsWith deep understanding in antibody-related realms and extensive project experience, Creative Biolabs offers a variety of references to help you learn more about our capacities and achievements, including infographic, flyer, case study, peer-reviewed publications, and all kinds of knowledge that can assist your projects. You are also welcome to contact us directly for more specific solutions.
HighlightsGet a real taste of Creative Biolabs, one of the most professional custom service providers in the world. We are committed to providing highly customized comprehensive solutions with the best quality to advance your projects.
HighlightsThe solute family 17 (anion/sugar carrier), member 5, is a protein encoded by the SLC17A5 gene in humans. It is also known as SLC17A5 or sialin. The lysogenic acid and free sialic acid separated from the lysosome are separated from the lysate, which is a necessary condition for the myelination of the normal central nervous system. The potential dependence of aspartate and glutamate membranes is absorbed into synaptic vesicles and synaptic-like microvesicles. At the same time, in the serosal membrane of salivary gland acinar cells, it can also act as an electrical 2NO3-/H+ transporter to regulate physiological nitrate effluent, and 25% of circulating nitrate ions are usually removed and secreted in saliva.
| Basic Information of SLC17A5 | |
| Protein Name | Sialin |
| Gene Name | SLC17A5 |
| Aliases | H(+)/sialic acid cotransporter, AST, Membrane glycoprotein HP59, Solute carrier family 17 member 5, Vesicular H(+)/Aspartate-glutamate cotransporter |
| Organism | Homo sapiens (Human) |
| UniProt ID | Q9NRA2 |
| Transmembrane Times | 12 |
| Length (aa) | 495 |
| Sequence | MRSPVRDLARNDGEESTDRTPLLPGAPRAEAAPVCCSARYNLAILAFFGFFIVYALRVNLSVALVDMVDSNTTLEDNRTSKACPEHSAPIKVHHNQTGKKYQWDAETQGWILGSFFYGYIITQIPGGYVASKIGGKMLLGFGILGTAVLTLFTPIAADLGVGPLIVLRALEGLGEGVTFPAMHAMWSSWAPPLERSKLLSISYAGAQLGTVISLPLSGIICYYMNWTYVFYFFGTIGIFWFLLWIWLVSDTPQKHKRISHYEKEYILSSLRNQLSSQKSVPWVPILKSLPLWAIVVAHFSYNWTFYTLLTLLPTYMKEILRFNVQENGFLSSLPYLGSWLCMILSGQAADNLRAKWNFSTLCVRRIFSLIGMIGPAVFLVAAGFIGCDYSLAVAFLTISTTLGGFCSSGFSINHLDIAPSYAGILLGITNTFATIPGMVGPVIAKSLTPDNTVGEWQTVFYIAAAINVFGAIFFTLFAKGEVQNWALNDHHGHRH |
SLC17A5 is a lysosomal membrane sialic acid transport protein encoded by the SLC17A5 gene on chromosome 6 in humans. The lack of protein leads to Salla disease and infant sialic acid storage disease (ISSD). The gene for HP59 is completely within its coding region, the Sialin gene SLC17A5. Infant sialic acid storage disease (ISSD) is a lysosomal storage disease characterized by the accumulation of covalent unchained (free) sialic acid in multiple tissues. ISSD and Salla disease (a major neurological disorder) is an allelic disorder caused by a recessive mutation in the lysosomal anionic monosaccharide transporter, SLC17A5.
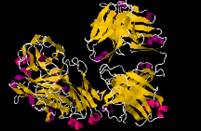
 Fig.1 The structure of SLC17A5 Protein.
Fig.1 The structure of SLC17A5 Protein.
This article reveals that combinations of neurological symptoms, visceral enlargement, malformation, and/or non-immune water corpus should be specifically tested for SASD in a timely manner to reduce diagnostic delay.
The clinical research shows that the patient is compound heterozygous of the c.908G>A (p.Trp303Ter) and the splicing mutation c.1259+5G>T (IVS9+5 G>T) in the SLC17A5 gene.
This study describes a novel pathogenic variant of SLC17A5, a translocation inserted in a patient with mild biochemical and clinical phenotypes.
These findings raise the possibility that in this population, a common founder mutation appears as hydrops. Furthermore, significant confirmation of the neuraminidase activity may be helpful in the clinical diagnosis of ISSD if confirmed in subsequent cases.
This study shows that SLC17A5 is most probably the major and possibly only vesicular transporter for NAAG and NAAG2 because ATP-dependent transport of both peptides has not been detected in vesicles isolated from sialin-deficient mice.
To obtain the soluble and functional target protein, the versatile Magic™ membrane protein production platform in Creative Biolabs enables many flexible options, from which you can always find a better match for your particular project. Aided by our versatile Magic™ anti-membrane protein antibody discovery platform, we also provide customized anti-SLC17A5 antibody development services.
Creative Biolabs' skillful scientists are glad to leverage our expertise and advanced technologies to help you with the member protein research. If you are interested, please feel free to contact us for more details.
All listed services and products are For Research Use Only. Do Not use in any diagnostic or therapeutic applications.
| USA:
Europe: Germany: |
|
|
Call us at: USA: UK: Germany: |
|
|
Fax:
|
|
| Email: info@creative-biolabs.com |