Creative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsCreative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsCreative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsCreative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsWith over a decade of experience in phage display technology, Creative Biolabs can provide a series of antibody or peptide libraries that are available for licensing or direct screening. These ready-to-use libraries are invaluable resources for isolating target-specific binders for various research, diagnostic or therapeutic applications.
HighlightsCreative Biolabs has established a broad range of platforms for developing novel antibodies or equivalents. These cutting-edge technologies enable our scientists to meet your demands from different aspects and tailor the most appropriate solution that contributes to the success of your projects.
HighlightsWith deep understanding in antibody-related realms and extensive project experience, Creative Biolabs offers a variety of references to help you learn more about our capacities and achievements, including infographic, flyer, case study, peer-reviewed publications, and all kinds of knowledge that can assist your projects. You are also welcome to contact us directly for more specific solutions.
HighlightsGet a real taste of Creative Biolabs, one of the most professional custom service providers in the world. We are committed to providing highly customized comprehensive solutions with the best quality to advance your projects.
HighlightsCreative Biolabs currently pushes out X-ray crystallography with rational designs, facilities, and expertise to assist scientists to crystallize biological macromolecules and determine their three-dimensional structures. Based on the technique of X-ray crystallography, Creative Biolabs can obtain interactions between attractive proteins from customers. This precise imaging technology not only reveals the best visualization of protein structures at the atomic level but also accelerates the comprehending of protein’s interaction and function.
X-ray crystallography is essentially a form of extreme high-resolution microscopy and represents the fact that X-ray is diffracted by crystal. With X-rays, the wavelength of electromagnetic radiation is around 0.1 nm to 1 Å long, which is just the right size to measure the distance between atoms in a molecule. Besides, the specificity of protein’s active and binding sites is completely dependent on the precise conformations of molecules. That’s why X-ray crystallography is chosen.
 Fig.1 An X-ray diffraction pattern of a crystallized enzyme.
Fig.1 An X-ray diffraction pattern of a crystallized enzyme.
Before running this assay, a purified protein sample at high concentration is crystallized. X-rays strike the single crystal exposed to a diffractometer, where the crystalline atoms cause a beam of incident rays to diffract into numerous specific directions. As the crystal is gradually rotated, the intensities and angles of diffractive beams are recorded and fed into a computer, which utilizes mathematical equation with Fourier transforms to calculate the position of each atom and then produces a 3D digital image of electron density. Lately, a model of the protein is constructed, adjusted and refined by the electron density maps and X-ray diffraction patterns.
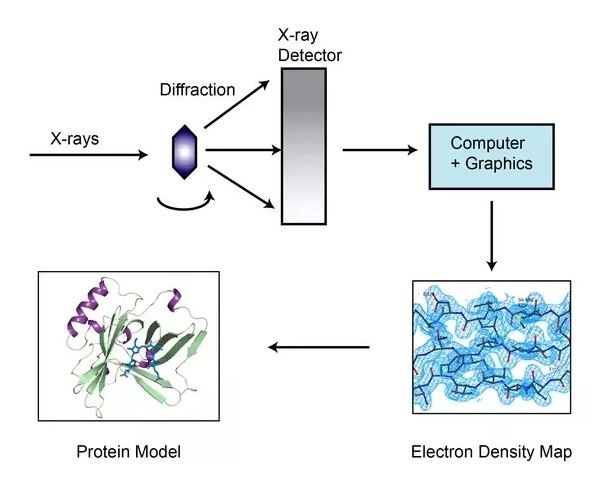 Fig.2 Overview of X-ray crystallographic method.
Fig.2 Overview of X-ray crystallographic method.
Crystallography can reliably provide the answer to structure related questions, from global folds to atomic details of bonding and the information from X-ray diffraction has enhanced the availability of protein structures and development on medicinal chemistry design greatly.
Functions of X-ray crystallography
Strengths on X-ray crystallography
The utilization of X-ray crystallography to measure protein structure has led to a significant breakthrough in the area of structural biochemistry and specifically provides extensional insights on protein-protein interactions within cellular processes.
X-ray crystallography provides a novel sight to explore interesting interactions of various proteins. The lab teams in Creative Biolabs have efficiently achieved high-resolution protein structure determination via X-ray crystallography to facilitate both structural biology and modern medical advancements. Armed with the results of X-ray crystallography services from Creative Biolabs, including sample purification, crystallization, and structure solution, combined with experienced operators and statistics analysis, clients will be guided to detail subsequent studies of relevant protein or complex crystals at structural, kinetic and thermodynamic aspects.
Other optional Protein-Protein Interaction (PPI) Assay services:
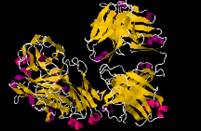
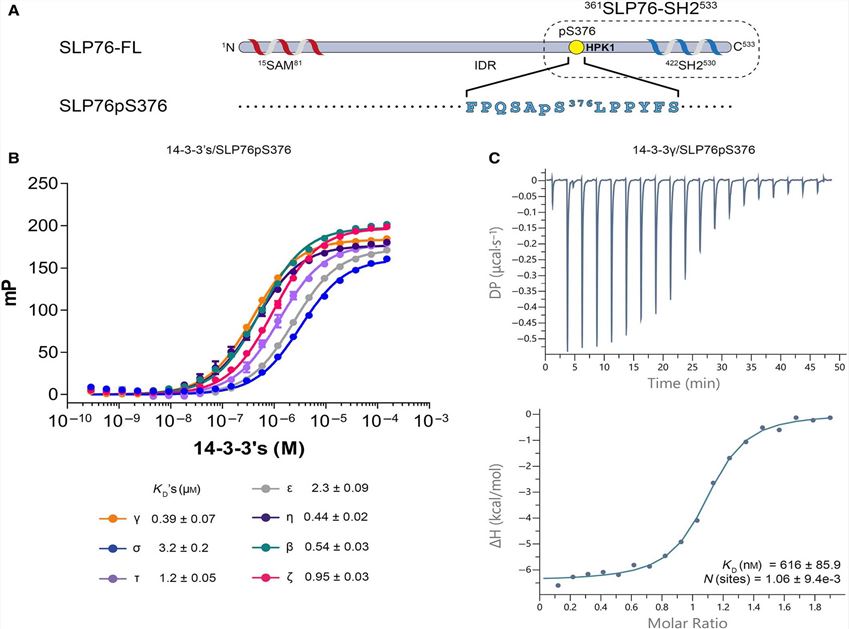 Fig. 3 Binding of the SLP76pS376 peptide to 14-3-3 proteins. (Lorenzo Soini, 2020)
Fig. 3 Binding of the SLP76pS376 peptide to 14-3-3 proteins. (Lorenzo Soini, 2020)
SLP76 is a connective protein containing the SH2 domain, which can coordinate the complex protein network downstream of the T cell receptor (TCR) and regulate the immune response. After phosphorylation on Ser376, SLP76 interacts with the 14-3-3 binding protein, resulting in its hydrolysis and degradation. This provides a negative feedback mechanism through which TCR signal transduction can be controlled. In order to further understand the 14-3-3/SLP76 protein-protein interaction (PPI), the researchers determined the high-resolution crystal structure of SLP76 synthetic peptides containing Ser376 and 14-3-3σ by X-ray crystallography. Its biophysical binding to 14-3-3 protein was characterized by fluorescence polarization and isothermal titration calorimetry. In addition, it was proven that the two recombinant SLP76 protein constructors also bind to 14-3-3.
This method involves co-crystallizing the proteins of interest together and then analyzing the resulting crystal with X-ray diffraction. The diffraction pattern provides detailed information about the spatial arrangement of atoms within the proteins, allowing researchers to deduce the structure of the protein complex. By understanding these interactions, scientists can gain insights into the function and mechanism of proteins in biological processes.
X-ray crystallography provides extremely high-resolution images of protein complexes, enabling precise mapping of the interaction sites and identification of involved amino acids. This level of detail can reveal the specific nature of binding sites and the conformational changes that occur upon interaction, which are crucial for understanding the biochemical basis of protein functions and their roles in cellular processes. Moreover, once a protein complex is successfully crystallized, X-ray crystallography can often be used to screen for potential inhibitors, which is valuable in drug development.
One of the primary challenges is the difficulty in obtaining high-quality crystals of protein complexes. Protein interactions can be transient or weak, making stabilization and crystallization challenging. Additionally, the proteins and their complexes may be sensitive to the conditions required for crystallization, such as pH and temperature, which can affect their natural conformation and binding properties. The process is also time-consuming and requires significant expertise in protein chemistry, crystallography, and data analysis.
X-ray crystallography is often compared to other techniques like nuclear magnetic resonance (NMR) spectroscopy, surface plasmon resonance (SPR), and isothermal titration calorimetry (ITC) for studying protein-protein interactions. While X-ray crystallography offers high-resolution structural details, it is limited to interactions that can be captured in a stable crystal form. NMR can provide information on protein interactions in solution, making it useful for observing dynamic changes and interactions that are difficult to crystallize. SPR and ITC are excellent for quantifying the binding affinity and kinetics of interactions, but they do not provide structural data. Thus, X-ray crystallography is often used in conjunction with these other methods to obtain a comprehensive understanding of protein interactions.
The time required to complete a protein-protein interaction assay by X-ray crystallography can vary significantly depending on several factors, such as the solubility and stability of the proteins, the complexity of the interactions, and the crystallization conditions. Typically, the process involves several stages, including protein purification, crystallization trials, optimization of crystals, data collection, and structure determination. This can take anywhere from several months to over a year. The crystallization step alone might require numerous trials and modifications to find the optimal conditions for forming high-quality crystals suitable for X-ray diffraction analysis.
X-ray crystallography provides unparalleled structural detail at the atomic level, which is essential for understanding the precise manner in which proteins interact. This method can reveal the exact locations of interaction sites, the orientation of proteins within complexes, and specific atomic interactions such as hydrogen bonds, salt bridges, and hydrophobic contacts. These details are crucial for designing drugs and inhibitors that can modulate protein function effectively. Other techniques might indicate that an interaction occurs and describe its strength or dynamics, but they generally cannot provide the same level of detailed structural information as X-ray crystallography.
For X-ray crystallography, the protein samples must be highly purified and stable under the conditions used for crystallization. They should also be available in sufficient quantity, as the process can require a significant amount of protein. Ideally, proteins used in crystallography should not have flexible regions, as these can hinder crystal formation. Modifications such as truncation of flexible regions, fusion to stability-enhancing partners, or specific point mutations might be necessary to achieve crystallization. In the context of protein-protein interactions, both interacting partners must be co-expressed or mixed in vitro in a stoichiometric ratio favorable for complex formation.
Use the resources in our library to help you understand your options and make critical decisions for your study.
All listed services and products are For Research Use Only. Do Not use in any diagnostic or therapeutic applications.
| USA:
Europe: Germany: |
|
|
Call us at: USA: UK: Germany: |
|
|
Fax:
|
|
| Email: info@creative-biolabs.com |






-a-versatile-tool-for-biomedical-research-and-diagnostics-1.jpg)